Manufacturers who wish to place medical devices on the market in the EU must comply with the European Medical Device Regulation MDR.
This Regulation (EU) 2017/745 on Medical Devices, as it is officially titled, also imposes requirements on notified bodies, distributors, importers, and health institutions such as hospitals.
… for beginners
If you are entirely new, download the free Starter Kit. It gives you an overview of the regulations, shows you the steps for “approval” of your medical device, and contains the MDR checklist in PDF and DOCX format for download.
… for advanced users
Take a deeper insight into the details on this page and the articles linked to it.
Benefit from the consolidated version of the MDR in English or German. It summarizes all changes to the MDR, including the transitional periods extended in March 2023. Internal links make it easier for you to navigate through the more than 170-page regulation.
… for IVDR manufacturers
Manufacturers of in-vitro diagnostic medical devices should read the article on the IVDR.
… for importers, distributors, and operators
Read the articles linked in chapter 1.b).
1. Overview of the MDR requirements
This article uses the abbreviation “MDR” as a synonym for Medical Device Regulation, EU Medical Device Regulation, and “Regulation (EU) 2017/745.“
1.1 Requirements for manufacturers and their medical devices
The requirements that the Medical Device Regulation places on manufacturers are extensive.
1.1.1 Cross-product requirements
| Requirements of the MDR and links to articles | Explanation |
| Quality Management System | All manufacturers require a QM system for developing, producing, and monitoring devices on the market, among other things. Except for class I devices, certification is usually also required. |
| Risk Management System | Risk management must ensure that the benefits are acceptable in view of the risks throughout the entire product life cycle. |
| Person Responsible for Regulatory Compliance (“PRRC”) | Manufacturers are obliged to employ a Person Responsible for Regulatory Compliance who ensures, for example, that the technical documentation is prepared in accordance with their own QM system and in compliance with the MDR. |
1.1.2 Product-specific requirements
| Requirements of the MDR and links to articles | Explanation |
| Device Classification | The manufacturer must determine the risk class for each device. |
| General Safety and Performance Requirements and Technical Documentation | The technical documentation must meet the requirements of Annex II and provide all evidence that the general requirements of Annex I are fulfilled. For this evidence, manufacturers are expected to apply harmonized standards and applicable common specifications. |
| Clinical Evaluation | As part of the clinical evaluation, the manufacturer must continuously check whether the devices’ safety, performance, and benefits are given. If the clinical data is not sufficient, a clinical investigation is necessary. |
| Unique Device Identification (UDI) | All medical devices must be given a Unique Device Identification, the UDI. This also means that the devices must be registered in EUDAMED. |
| Labeling | The MDR precisely defines requirements for instructions for use, other accompanying materials, and labeling such as product labels and packaging. |
| Conformity Assessment | Depending on the risk class of the device, manufacturers must undergo a conformity assessment procedure. Except for class I devices, they must involve a notified body. If successful, manufacturers must prepare a declaration of conformity and affix the CE mark. |
| Post-Market Surveillance and Vigilance | Manufacturers are obliged to monitor their medical devices on the market throughout their entire lifecycle , continuously collect respective data , and take corrective actions if necessary. If there are negative trends related to incidents, they must inform the authorities. |
1.2 Requirements for other stakeholders (distributors, importers, etc.)
Not only medical device manufacturers must comply with the Medical Device Regulation, but also other stakeholders:
- Requirements for distributors
- Requirements for importers
- Information for hospitals and other operators
- Requirements for manufacturers of specific devices without an intended medical purpose (Annex XVI devices)
This article on the differences between the MDR and MDD is beneficial for manufacturers of legacy devices.
This article on the transitional periods is also essential for these manufacturers.
2. Organization and structure of the MDR
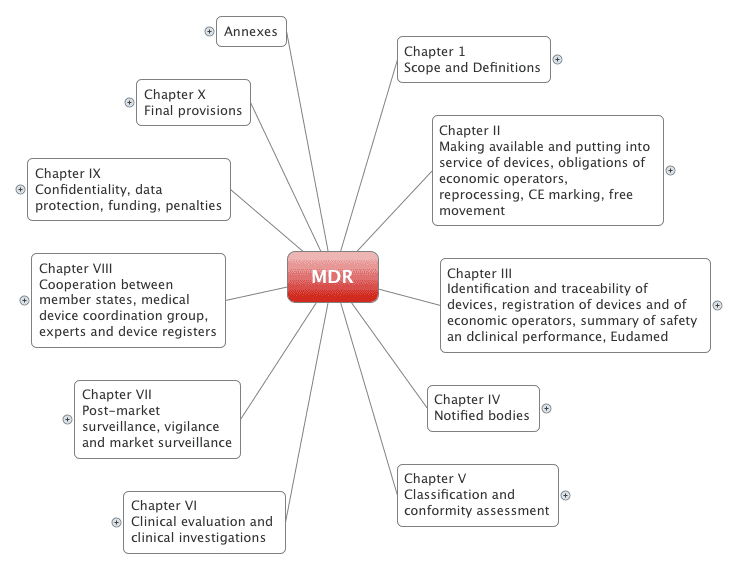
The Medical Device Regulation comprises 123 articles divided into 10 chapters. It also has 17 annexes.
2.1 The chapters
The Medical Device Regulation (MDR) has been completely restructured compared to the old Medical Device Directive (MDD):
| Chapter number With links to MDR | Title / Content / Requirements With links to articles |
| I | Scope of application and definitions |
| II | The most important requirements for manufacturers, distributors, importers, and member states. Many references to articles in other chapters and the annexes |
| III | Traceability of devices, especially requirements for UDI |
| IV | Requirements for notified bodies |
| V | Classification and conformity assessment procedures |
| VI | Clinical evaluations and clinical investigations |
| VII | Post-market surveillance, market surveillance, reporting |
| VIII | Cooperation between member states, MDCG, and other experts |
| IX | Confidentiality, data protection, penalties |
| X | Transitional periods and more |
2.2 The annexes
The EU Medical Device Regulation has 17 annexes.
| Annex number With links to the MDR | Title / Content / Requirements With links to further information |
| I | General safety and performance requirements |
| II | Technical documentation |
| III | Technical documentation for post-market surveillance |
| IV | EU Declaration of Conformity |
| V | CE marking |
| VI | UDI, EUDAMED |
| VII | Requirements for notified bodies |
| VIII | Classification rules |
| IX to XI | Conformity assessment procedure: – Complete QM system – Type examination – Product conformity verification |
| XII | Certificates from notified bodies |
| XIII | Custom-made devices |
| XIV | Clinical evaluation |
| XV | Clinical investigation |
| XVI | Devices without an intended medical purpose |
| XVII | Correlation table |
3. Further articles and links
3.1 Further articles
The EU has postponed the transitional periods for the application of the MDR several times. The rules are so complex that an article on the transitional periods is helpful.
Further explanations and requirements have been published by the Medical Device Coordination Group (MDCG).
There are different requirements for devices without an intended medical purpose (“Annex XVI devices”).
3.2 Further links
- EU 2027/745 (original text)
- English (HTML) linked version of the Johner Institute (consolidated)
- English (HTML) Original version of the EU
- English (PDF) Original version of the EU
- German (HTML) linked version of the Johner Institute (consolidated)
- German (HTML) Original version of the EU
- German (PDF) Original version of the EU
- Corrigenda
- 1st Corrigendum: REGULATION (EU) 2020/561 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 23 April 2020
- 2nd Corrigendum: COMMISSION DELEGATED REGULATION (EU) 2023/502 of 1 December 2022
- 3rd Corrigendum: REGULATION (EU) 2023/607 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 15 March 2023
- 4th Corrigendum: REGULATION (EU) 2024/1860 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 13 June 2024
- Other
4. Background information on the EU Medical Device Regulation
4.1 How the EU regulations came about
It is often claimed that the breast implant scandal triggered the revision of medical device legislation. However, most stakeholders now deny this. Therefore, it is largely unclear who initiated the new regulation and for what reason.
4.2 Difference between EU regulations and EU directives
Like all EU directives, the “old” EU medical device directives had to be transposed into national laws and national regulations in order to become law. In Germany, these were the “Medizinproduktegesetz” (MPG) and ordinances such as the Medical Devices Operator Regulation (still valid) and the Medical Devices Safety Plan Regulation (now invalid).
The EU regulations (here: MDR, IVDR) have a direct legal character. National laws only supplement these regulations, such as the MPDG and others in Germany, with penal provisions and the definition of responsibilities of the national competent authorities.
4.3 Criticism of the EU Medical Device Regulation
The criticism of the EU Medical Device Regulation is massive. Efforts and costs have multiplied for manufacturers. The time taken for the “approval procedures” has also increased.
As a result, competitiveness, innovation, and the supply of medical devices have been hampered.
These consequences were already feared in 2020.
For future regulation, it is essential to understand the system – a task for regulatory science.
5. Conclusion and summary
The EU Medical Device Regulation MDR is a comprehensive piece of legislation that poses significant challenges for all stakeholders (medical device manufacturers, notified bodies, distributors, importers, hospitals). It is not apparent that the MDR will improve medical devices’ safety, performance, and effectiveness. However, the supply of medical devices is at risk.
Manufacturers have little choice but to deal intensively with this legislation and comply with its requirements. These increased requirements are not limited to the “approval” (pre-market phase) but also explicitly affect the post-market phase (post-market surveillance, vigilance).
Support with the implementation of the MDR
Free offers
Do you still have questions about the MDR and its implementation? You can get answers in our free micro-consulting.
Download the free Starter Kit. It gives you an overview of the regulatory landscape and contains the MDR checklist as a PDF and in DOCX format!
Videos and e-learning
The video training courses at our Medical Device University show you step-by-step how to create your technical documentation and QM system in a lean, fast, and MDR-compliant way. Over 100 templates and sample documents are available for download. In this way, you create the prerequisites for approving your devices quickly and safely and launching them on the market.
Product testing
The experts at the Johner Institute can help you test your devices:
- Usability tests
- Penetration tests
- Electrical safety and EMC tests
- Biocompatibility tests
- Clinical evaluations and investigations
Consulting
Benefit from the know-how of our regulatory affairs experts to
- define your regulatory strategy and classify devices,
- create technical documentation (TD),
- set up QM systems (QMS),
- review TD and QMS and prepare for audits and reviews, and
- simplify post-market surveillance.
Contact us immediately so we can clarify together how you can quickly and efficiently meet the regulatory requirements of the MDR and bring your devices safely to market.
Change history (as of May 2021)
- 2025-02-04: Empty note box filled in at the beginning of the article, various synonyms in the article used
- 2024-08-27: Links to corrigenda corrected and links grouped hierarchically in chapter 3.2
- 2024-01-18: Chapter numbering changed
- 2023-04-18: Article completely rewritten
- 2021-10-10: Section 7.e) (The consequences were not considered and are not understood) added
- 2021-07-26: Link to new corrigendum added
- 2021-05-24: New article on the declaration of conformity linked
