The EU Medical Devices Regulation (MDR) regulates not only medical devices but also devices without an intended medical purpose, e.g., liposuction devices, breast implants, and colored contact lenses.
In December 2022 – four and a half years after the MDR was published – the EU regulated the necessary details with two Commission Implementing Regulations (2022/2346 and 2022/2347).
For manufacturers of these devices the following sources are particularly relevant:
- MDR Annex XVI
- Common Specifications: Commission Implementing Regulation (EU) 2022/2346 in EU Official Journal (EN)
- Classification: Commission Implementing Regulation (EU) 2022/2347 on the reclassification of rules 9 and 10 for Annex XVI devices (EN)
This article provides manufacturers of devices without an intended medical purpose (Annex XVI devices) with an overview of the regulatory requirements they have to comply with and offers practical tips for their implementation.
1. Devices subject to Annex XVI
Annex XVI of the MDR lists six groups of devices.
| # | product group | examples |
| 1 | contact lenses or other items intended to be introduced into or onto the eye | coloured contact lenses |
| 2 | devices intended to be totally or partially introduced into the human body through surgically invasive means for the purpose of modifying the anatomy or fixation of body parts with the exception of tattooing devices and piercings | subdermal implants such as horn implants, breast implants |
| 3 | substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction, excluding those for tattooing | dermal fillers, such as hyaluronic acid injections |
| 4 | equipment intended to be used to reduce, remove, or destroy adipose tissue, such as equipment for liposuction, lipolysis, or lipoplasty | body shaping devices, e.g., for liposuction |
| 5 | high intensity electromagnetic radiation (e.g., infra-red, visible light, and ultra-violet) emitting equipment intended for use on the human body, including coherent and non-coherent sources, monochromatic and broad spectrum, such as lasers and intense pulsed light equipment, for skin resurfacing, tattoo, or hair removal, or other skin treatment | IPL equipment for hair removal or skin rejuvenation; does NOT apply to sunbeds |
| 6 | equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain | non-invasive equipment for magnetic or electrical stimulation of the brain (without a specific medical purpose) |
The EU Commission may extend this list in the future and add new product groups by means of delegated acts (Implementing Regulations). However, this requires these devices to be similar to corresponding medical devices based on the risk profile or features or their functionality.
2. Requirements for devices without an intended medical purpose
a) Almost the same requirements apply as for medical devices
The abridged version is: Devices without an intended medical purpose are subject to (almost) the same requirements as “regular” medical devices.
The MDR even groups medical devices, accessories, and devices without an intended medical purpose under the term “devices” and does not differentiate between them (with a few exceptions).
Hence, the requirements under MDR Article 10 also apply. In other words: Manufacturers must:
- Establish and implement a quality management system (including a risk management system)
- Conduct a clinical evaluation
- Compile technical documentation in accordance with MDR Annexes II and III
- Undergo a conformity assessment procedure and issue a declaration of conformity
- Comply with the UDI requirements
- Establish a post-market surveillance system
- Comply with the reporting requirements (vigilance)
- Make provisions for liability arising from defective devices, e.g., arrange for adequate insurance coverage
The MDR requires that devices comply with the general safety and performance requirements that apply to them, as set out in Annex I.
b) There are, however, also differences
Medical devices and Annex XVI devices differ in some of their requirements.
The Common Specifications describe these differences mainly in the following annexes:
| annex | devices | note |
| Annex I | all devices | The annex mainly details the requirements for risk management and “information for safety,” e.g., instructions for use and labeling. |
| Annex II | contact lenses | This annex details the additional specific risk management and information for safety requirements for this product class. |
| Annex III | devices introduced into the body through surgically invasive procedures, e.g., horn implants and breast implants | ditto |
| Annex IV | facial or other dermal or mucosal fillers intended for subcutaneous, submucosal, or intradermal injection or other modes of delivery | ditto |
| Annex V | devices to reduce, remove, or destroy adipose tissue | ditto |
| Annex VI | devices intended for use on the human body that emit high-intensity electromagnetic radiation, such as devices used for skin resurfacing, tattoo removal, hair removal, or other forms of skin treatment | ditto In addition, this Annex defines the terms “device for professional use” and “device for home use.” |
| Annex VII | equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain | This annex details the additional specific risk management and information for safety requirements for this product class. |
i) Clinical evaluation without demonstrating benefit
It would be difficult to demonstrate clinical benefit for devices without an intended medical purpose, as is required for medical devices. This is why clinical evaluation under Article 61 “only” needs to demonstrate the safety and performance of these devices but not the clinical benefit (see Article 61(9)).
Some devices may be considered both a medical device and an Annex XVI device. One such example is breast implants for medical and aesthetic purposes. In this case, clinical benefit must also be demonstrated.
ii) General requirement for clinical investigations
The general requirement for clinical investigations for Annex XVI devices is interesting. This can only be sidestepped if sufficient clinical data is available and has been assessed for an “analogous” medical device.
MDR uses the term “analogous” and not “equivalent.” There is no definition of the term “analogous” in the MDR. This is provided by the Common Specifications:
“An analogous device with a medical purpose shall be understood as the same device with a medical purpose or a medical device for which equivalence to the same device with a medical purpose has been demonstrated by the manufacturer in accordance with Section 3 of Annex XIV to Regulation (EU) 2017/745 of the European Parliament and of the Council.”
iii) Risk acceptance criteria that are not based on benefit
Another difference concerns the risk-benefit ratio required in Annex I (see Annex I, Chapter I, 9.). In the absence of a medical benefit, manufacturers must determine risk policy and acceptance on a different basis.
Again, a look at the Common Specifications will help:
“If the undesirable side-effects are of transient nature and do not require medical or surgical intervention to prevent life-threatening illness or permanent impairment of a body function or permanent damage to a body structure, residual risks may be considered as being acceptable. If one or more of the conditions laid down in this Section is not met, the manufacturer shall provide a justification explaining the reasons for the acceptability of the risks.”
Commission Implementing Regulation (EU) 2022/2346
Thus, risk acceptance must be based on severity rather than probability of potential harm. This is an unfortunate requirement because severe (e.g., irreversible) harm can always occur in a very unlikely case.
iv) Product-specific requirements
The real difference is in the product-group specific requirements that the Common Specifications mandate, the “common specifications” in Annexes I to VII of the Commission Implementing Regulation (EU) 2022/2346.
These requirements are based on the general safety and performance requirements in MDR Annex I, such as risk management or labeling, but are product-specific.
Risk analysis
Annex I of the act applies to all Annex XVI devices and describes, among other things, the required risk management process. This is very closely aligned with ISO 14971.
It names product-specific hazards that manufacturers must assess.
Example: Section 2 devices such as subdermal implants (Annex III of the act): Specific hazards, e.g., microbiological contamination, implant failure and movement, implant shrinkage, and wrinkling, discomfort or pain, etc.
The authors also classify discomfort and pain as hazards. It would be better to speak of harms.
Risk control
The Common Specifications not only identify hazards that manufacturers must consider but also specific risk control measures. For example, they require that implants be sterile and non-pyrogenic; that long-term data be collected to assess the presence of non-degradable materials; or that manufacturers provide training on implantation and safe use of the product.
Labeling
Manufacturers must clearly indicate the non-medical purpose in the labeling. In implants, the label must state that “the devices must not be used on persons under 18 years of age.“
3. Conformity assessment of devices without an intended medical purpose
a) Conformity assessment procedure
The MDR requires the same conformity assessment procedures for Annex XVI devices as for medical devices. Even for devices without an intended medical purpose, the class of the product determines which conformity assessment procedure manufacturers are allowed to use.
b) Classification
In MDR Annex XVI devices, this classification is also governed by the rules set out in MDR Annex VIII. However, not all rules can be applied. For example, rules 9 and 10 (which are the rules for active therapeutic and diagnostic devices) assume a medical purpose.
As a result, another Commission Implementing Regulation (2022/2347) was adopted for the reclassification of relevant devices at the same time as the Common Specifications:
| product class | detailed in Section | class |
| body shaping devices | 4. | IIb |
| devices for skin rejuvenation, hair removal, etc. | 5. | hair removal: class IIa, otherwise IIb |
| devices for transcranial brain stimulation | 6. | III |
4. Transitional periods for Annex XVI devices
The Commission Implementing Regulations were not published until four and a half years after the MDR. It is possible that this fact, as well as experience with the transitional periods for medical devices, has resulted in sometimes generous transitional periods.
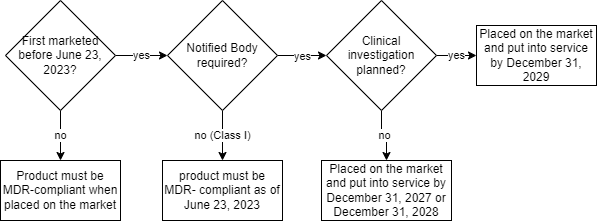
Which transitional periods apply depends on the setting in question:
| # | setting | transitional period | conditions |
| 1 | clinical investigation in progress or planned AND involvement of a notified body required (> class I) | placing on the market and putting into service by December 31, 2029 | – first placing on the market before June 22, 2023 – marketing continues in accordance with the pre-MDR legislation for the respective product group – no significant changes to the design or intended purpose – complete clinical investigation application including regulatory confirmation by June 22, 2024 – start of clinical investigation no later than December 23, 2024 – signed conformity assessment contract with notified body before January 1, 2028 |
| 2 | NO clinical investigation planned AND involvement of a notified body required (> class I) | placed on the market and put into service by December 31, 2028 | – first placing on the market before June 22, 2023 – marketing continues in accordance with the pre-MDR legislation for the respective product group – no significant changes to the design or intended purpose – signed conformity assessment contract with notified body before January 1, 2027 |
| 3 | valid current MDD certificate (expired between May 26, 2021 and March 20, 2023) | also after expiry of the certificate until December 31, 2027 (class III and specific class IIb implantable devices) or December 31, 2028 (others) | – no signed contract for MDR certification with notified bodies before certificate expiry AND no “special authorization” according to Article 59 or 97 of the MDR – marketing continues in accordance with the MDD requirements – no significant changes to the design or intended purpose – no unacceptable risk – QMS according to Article 10(9) MDR at the latest by May 26, 2024 – application for conformity assessment MDR by May 26, 2024 at the latest – signed contract for conformity assessment MDR by September 26, 2024 at the latest |
| 4 | no involvement of a notified body required (class I) | placing on the market and putting into service by June 22, 2023 | – declaration of conformity according to MDR by June 22, 2023 at the latest |
The transitional periods thus depend on:
- date when the product was first placed on the market
- product class
- necessity of a clinical investigation
5. Recommendations on how to proceed
Many non-medical device manufacturers will be heavily taxed, and some overwhelmed, by the complexity and quantity of regulatory requirements.
Depending on their level of expertise, they should follow the steps below:
- Become familiar with the regulatory requirements. This includes, above all, reviewing the sources listed at the beginning of this article.
- Research harmonized standards that can be used to demonstrate compliance with these requirements.
- Identify the class of the product.
- Find out if a clinical investigation is mandatory.
- Identify the transitional periods.
- Find a notified body for devices of class IIa and higher.
- Establish a quality management system and have it certified, if needed.
- Compile the conformity documents.
- Plan and conduct the clinical investigation, if applicable.
- Declare the conformity of your devices and market them.
Johner Institute assists Annex XVI manufacturers every step of the way. Feel free to contact us!
6. Conclusion and summary
Lawmakers have set very high requirements for devices without an intended medical purpose according to MDR Annex XVI. This will most likely result in a massive “market shakeout.” Presumably, this is precisely what the lawmakers were aiming for.
Regrettably, it has taken lawmakers well over four years to draft Commission Implementing Regulations that do not fully live up to a rigorous standard.
Compliance with these new requirements has now become the responsibility of the authorities. These are faced with the task of monitoring the market, for example by scouring internet stores and visiting pertinent trade shows. The goal of the legislation will not be achieved without this enforcement.
Change history
- 2023-06-22: Article adapted to the new transitional periods of Implementing Regulation 2023/1194 of June 20, 2023
