Since May 25, 2017, the In Vitro Diagnostic Medical Device Regulation (IVDR) has been in force, which has now replaced the previous IVD Directive (98/79/EC, IVDD). There are significant differences between the IVDR and IVDD.
This article presents these differences. In doing so, it provides manufacturers who placed their devices under the EU directive (IVDD) on the market with an overview of the “gaps” that need to be closed. It also helps health institutions that have not yet been affected by the IVDR, such as medical laboratories.
Please also refer to the main article on the IVDR as well as to the article on transition periods.
1. Differences between the IVDR and IVDD: How they occurred
a) Trigger
It is widely rumored that the breast implant scandal was the catalyst for the revision of medical device legislation. However, most of those involved now deny this. It is, therefore, largely unclear who initiated the new regulation and for what reason.
b) Differences regarding the liability
The EU Medical Device Regulations (MDR, IVDR) replace the Medical Device Directives (MDD, AIMD, IVDD).
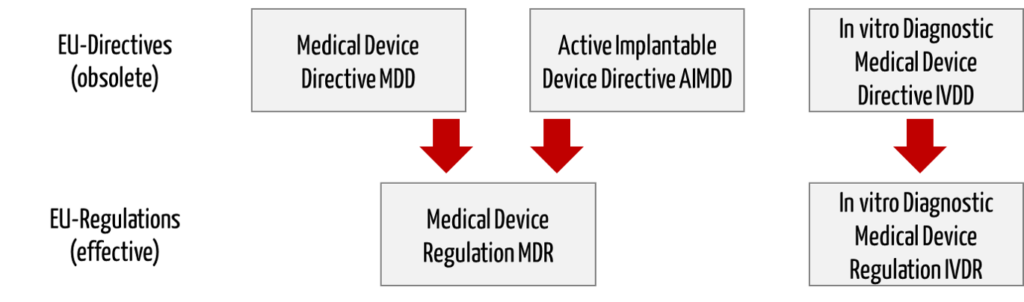
Directive 98/79/EC on in vitro diagnostic medical devices (IVDD) will be replaced by a separate regulation (In vitro Diagnostic Medical Device Regulation (IVDR), number 2017/746).
Read more about the Medical Devices Regulation, MDR.
c) Scope and structure of the IVDR
The EU regulation is divided into ten chapters and 14 annexes. While the IVDD contained 24 articles, the IVDR comprises 113 articles, indicating that the IVDR has been written from scratch.
The main article on the IVDR presents the chapter structure and the individual chapters.
2. What the IVDR has not changed
The IVDR has retained some concepts. For example, the regulation continues to recognize “essential requirements,” which are now called general safety and performance requirements.
The performance parameters to be demonstrated also hardly differ between the IVDD (Annex I, Section 3) and the IVDR (Annex I, 9.1.). Only a better distinction is made between analytical performance characteristics (Annex I, 9.1.a) and clinical performance characteristics (Annex I, 9.1.b).
Manufacturers still have to demonstrate that they and their devices meet these requirements by means of a conformity assessment procedure. The IVDR has also not changed the fact that conformity must be expressed by a CE mark.
3. Important changes due to the IVDR
a) Differences in the scope of application
In contrast to the IVDD, the IVDR regulates devices manufactured and used only within health institutions, the so-called in-house IVDs (also known as Lab-Developed Tests or LDTs). This means that medical laboratories must comply with the requirements of the IVDR.
b) Differences in classification and approval
Classification
For the classification of IVDs, the IVDR introduces risk-based classification rules. (Under the IVDD, classification was based on a list system.) For each device, an IVD manufacturer must go through all of the implementing and classification rules in Annex VIII to assign a device to one of the four classes:
- Class D: High individual and high public risk, life-threatening risk if the output is incorrect for multiple individuals, e.g., IVD for transfusion medicine or for determination of life-threatening and simultaneously highly infectious diseases
- Class C: High individual or moderate public risk, life-threatening risk if the output is incorrect for one individual, e.g., genetic testing, test to determine drug levels, infectious diseases, or congenital diseases in fetuses or embryos. Most devices for self-testing (by lay persons) also fall into this class.
- Class B: Moderate individual or low public risk, no life-threatening risk if the output is incorrect. Class B is the “default class” for IVDs that do not fall under the other classification rules.
- Class A: Low individual and low public risk devices without patient-specific information. This class includes non-critical devices such as wash solutions, culture media, and specimen receptacles.
The IVDR distinguishes other types of in vitro diagnostic medical devices:
- Devices for near-patient testing. These are classified on their own, taking into account all classification rules.
- Devices for self-testing (by patients/lay persons) always fall into class C, with some explicitly listed exceptions.
- Companion Diagnostics (CDx) essential for the safe and effective use of a corresponding medicinal product are assigned to class C.
Conformity assessment procedure
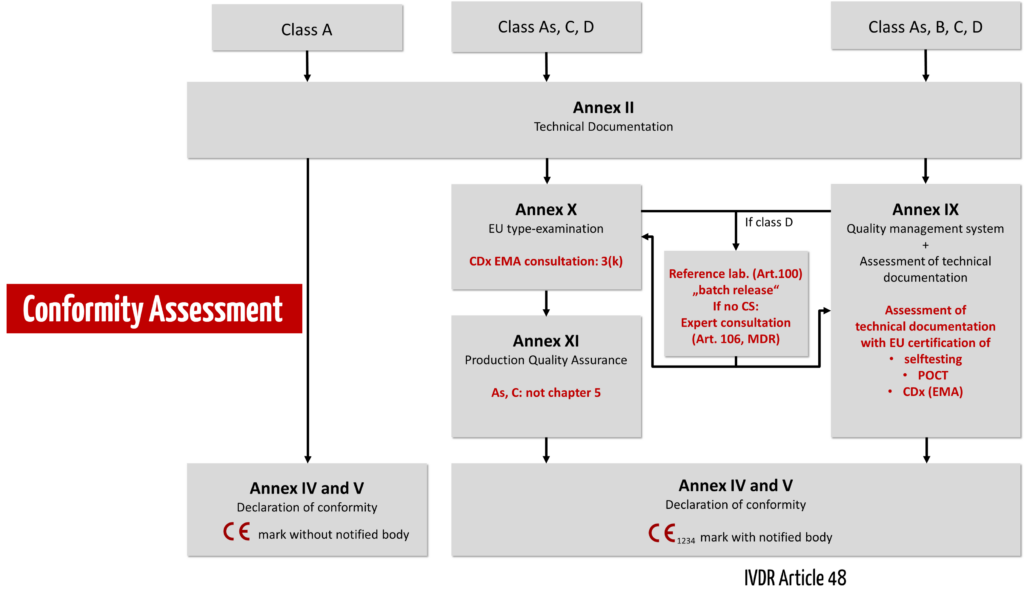
Under the IVDR, conformity assessment is no longer carried out by the manufacturer himself but by a notified body. Only manufacturers of non-sterile class A devices may still declare conformity themselves. There is still no central regulatory authority (like the EMA for medicinal products).
According to the general obligations for manufacturers according to Article 10 of the IVDR, IVD manufacturers must implement and enforce a quality management system that meets the requirements of the IVDR, Article 10, and ISO 13485 as harmonized standard. For all devices, regardless of their class, manufacturers must prepare product-specific technical documentation according to Annex II as well as technical documentation on post-market surveillance according to Annex III.
The IVDR essentially distinguishes between three conformity assessment procedures:
- Self-declaration of conformity by the manufacturer for class A devices without requirements for sterility.
- Conformity assessment based on the QM system and the evaluation of the technical documentation (Annex IX). The IVDR differentiates in the review of the technical documentation by a notified body whether this must be done per product category (class B), per product group (class C), or per device (class D). 3.
- Alternatively, manufacturers may choose a conformity assessment procedure based on type examination (Annex X) or production quality assurance (Annex XI).
For devices intended as near-patient testing (Point-of-Care Tests, POCT) or for self-testing by lay persons, the IVDR imposes specific requirements, as it does for Companion Diagnostics (CDx). Class D devices must involve reference laboratories and, if Common Specifications (CS) are not available, expert panels.
Common Specifications
Manufacturers should continue to use harmonized standards. The EU Commission also reserves the right to define so-called Common Specifications (CS) with which manufacturers must comply. These serve to provide a uniform specification of safety and performance requirements, particularly for high-risk products.
c) Technical documentation
Manufacturers must describe very precisely,
- what the device is intended to do,
- how they will ensure that the general safety and performance requirements are fulfilled, and
- that there are no unacceptable risks.
Intended use
The first essential activity includes defining a precise intended purpose. The information that must be specified can be found in Annex II, Section 1.1 and Annex I, Section 20.4.1.c – a valuable source for both IVD manufacturers and users.
Manufacturers and also medical laboratories developing and using in-house IVDs quickly realize that without a precise formulation of the intended purpose, neither a targeted regulatory strategy is possible nor the scope for the performance evaluation can be determined.
Performance evaluation
However, it is the performance evaluation that the IVDR focuses on. It describes the requirements for performance evaluation documentation in much greater detail than the IVDD. It devotes attention to the three elements of performance evaluation: scientific validity, analytical performance, and clinical performance (Annex XIII, Part A).
Post-market follow-up
The IVDR indicates that performance – as well as risks – must be monitored throughout the life cycle of a device and that performance evaluation is not a one-time thing. Therefore, the regulation requires Post-Market Performance Follow-up (PMPF) after placing a device on the market, in accordance with Annex XIII, Part B.
That performance evaluation and PMPF have a special place in the conformity assessment of an IVD is also demonstrated by the focus of the notified bodies during the assessment of the technical documentation. This, in turn, is in line with the requirement that only devices that can demonstrate their clinical benefit are on the market. And in the case of IVDs, this lies in particular in “providing accurate medical information on patients” (IVDR, preamble (64)).
The term “accurate” refers to, i.e., analytical and clinical accuracy including trueness and precision of the results.
d) Differences in product requirements
Risk management
Whereas the requirements for risk management previously originated more from ISO 14971, the IVDR now defines them in more detail in Annex I.
Performance studies
Comparable to the MDR, the requirements for documenting and conducting (clinical) performance studies have multiplied. This is evidenced by Article 57 and Annex XIII, Section 2, or Article 58 et seq. and Annex XIV. The latter impose further requirements on certain performance studies that are associated with a risk to the patient.
In 2019, the ISO 20916 was published as standard defining the special requirements for performance studies with IVDs. The standard is referenced in the preamble of the IVDR.
Read this article on sample size planning for performance studies with IVD.
Labeling
Page by page, the IVDR describes in Paragraph 20 of Annex I what manufacturers must consider regarding labeling (label, instructions for use, warnings, and other information).
Software
The IVDD addressed software very poorly. For example, it did not formulate any concrete requirements for the software life cycle. The IVDR changes that:
- Software must be developed in accordance with software lifecycle processes, including verification and validation.
- IT security is now an explicit requirement.
- Requirements for interoperability must be specified in detail by manufacturers, and compliance must be verified. Speaking of which, the new ISO 13485:2016 also requires this in the current version.
- Risk management must look at risks due to interoperability issues.
- The runtime environment, such as the operating system and hardware, must be defined by manufacturers.
- The regulation even addresses mobile platforms, where screen sizes and the use environment must be specified.
- Manufacturers must provide documentation that identifies components, algorithms, and technologies.
- The software itself is subject to Unique Device Identification (UDI).
- Personnel must also be competent with regard to software.
Overall, these are not surprising requirements. Rather, they reflect what IEC 62304 and, in part, ISO 13485 specify more concretely.
UDI system
As with other medical devices, manufacturers are required to clearly label the devices using a UDI and to upload information in EUDAMED.
e) Differences concerning the manufacturers
Quality management system
Every(!) manufacturer of an IVD medical device must have a quality management system (QM system). This applies regardless of the conformity assessment procedure. However, the IVDR only requires an assessment of the QM system by a notified body for the conformity assessment procedures according to Annex IX and XI.
The requirements for the quality management system are comprehensive and specified both via the obligations as a manufacturer in Article 10 of the IVDR and by ISO 13485 as a harmonized standard. The QM system must address, among other things:
- Organization, processes, responsibilities (including those of management)
- Strategy to ensure compliance
- Specification of requirements
- Resource management
- Supplier qualification
- Risk management
- Evaluating the performance of devices
- Development
- Production
- Maintenance
- Post-market surveillance
- UDI
- CAPA
Once you have described and implemented all these procedures, it is not a big step to compliance with ISO 13485:2016.
The IVDR requires notified bodies to perform an unannounced audit at least every five years for a chosen conformity assessment procedure according to Annex IX and Annex XI.
Post-market surveillance
Manufacturers must precisely plan and perform post-market surveillance (as part of the QM system). The IVDR describes the requirements in detail, including in Article 78 ff. and Annex III.
Personnel
As in ISO 13485:2016, the IVDR places increased emphasis on adequate qualification of personnel. The Person Responsible for Regulatory Compliance is a new, explicitly required role.
4. How to manage differences between the IVDR and IVDD
To overcome the differences in the requirements of IVDR and IVDD, you, as a manufacturer, should take the following steps:
- Ensure that the QM system meets the requirements of the IVDR and ISO 13485:2016.
- Formulate a precise intended purpose according to Annex II, Section 1.1. or Annex I, Section 20.4.1.c) and document it. It must be supported by appropriate performance data. Also, do not use different wording for marketing.
- Determine the classification of your devices.
- Create a gap analysis for the technical documentation and the post-market system.
- Close the gaps found in the process.
- Determine your UDI strategy together with development, logistics, and production.
- Coordinate the time and implementation plan with your notified body.
5. Conclusion
The IVDR is significantly more comprehensive than the IVDD. This is mainly due to the more specific requirements for documentation in the IVDR.
Essential differences are the risk-based classification rules and the conformity assessment by notified bodies for all class B, C, and D devices.
That the IVDR will lead to greater safety for patients, users, and third parties through the necessary clinical evidence and performance evaluation of devices cannot be doubted. However, it is questionable whether the IVDR, just like the MDR, could not have managed with less bureaucracy, as this has the effect of inhibiting innovation.
Do you need assistance with
- the precise formulation of your intended purpose,
- compiling lean technical documentation, or
- deriving a product-specific performance evaluation strategy?
Then get in touch right away. Here you will receive fast and uncomplicated help.
