What MDR and IVDR confuse and why you should not talk about CAPA.
The FDA (in 21 CFR part 820 – QSR) and ISO 13485 differentiate between
- corrective actions,
- preventive actions, and
- corrections.
Unfortunately, the MDR and IVDR do not clearly differentiate between these concepts. Some manufacturers also believe they can combine corrective and preventive actions into one single CAPA process. But this is just as imprecise as the lack of distinction between corrections and corrective actions.
Therefore, it is misleading to speak of a “CAPA process.” Statements such as “The CAPA process begins with identifying the problem” or “The process begins when a nonconformity or error occurs” also demonstrate an inadequate understanding!
Manufacturers generally need several processes to meet regulatory requirements.
This article defines the terms and helps you avoid deviations in audits and even illegal marketing of devices caused by this confusing terminology. It lists the regulatory requirements and uses examples to explain how to differentiate between the pairs “corrective action” and “correction” as well as “corrective action” and “preventive action.”
1. Correction
Definition
ISO 9000 defines the term correction as follows:
“action to eliminate a detected nonconformity”
Source: ISO 9000:2015 3.12.2
Examples
Examples of corrections are:
- Shortening a component that is too long
- Fixing a software bug
- Classifying a medical device in the right class
2. Corrective action
a) Corrective actions in ISO 9000 and ISO 13485
Definition
ISO 9000 defines the term corrective actions as follows:
“action to eliminate the cause of a nonconformity and to prevent recurrence”
Source: ISO 9001:2015 3.12.2
Therefore, the aim of a corrective action is not to eliminate nonconformities but to eliminate the causes of nonconformities that have already occurred and to ensure that such nonconformities do not occur again.
Colloquially, actions intended to ensure that a nonconformity does not occur again are often referred to as preventive actions. However, according to the definition, this is not a preventive action.
Examples of corrective actions
- Changing an incorrect setting on a production machine, e.g., CNC milling machine, so that the component is in the correct length in the future
- Revising the coding guidelines after a software error to ensure the error (probably) does not re-occur
- Establishing a new data protection strategy after a data loss
- After an incorrect classification, making further training mandatory for persons before they classify devices
- Automating the final inspection so that it is no longer possible to forget to document the results of the inspection
b) Corrective actions according to the MDR and IVDR
Unfortunately, MDR and IVDR have not adopted the definition of corrective action from ISO 9000 and ISO 13485:
“action taken to eliminate the cause of a potential or actual non-conformity or other undesirable situation;”
Source: MDR Article 2
This definition is very unfortunate because it mixes the elimination of the cause of a potential nonconformity and the elimination of the cause of an existing nonconformity. Elimination of a potential nonconformity is usually considered a preventive action.
Regrettably, the MDR and IVDR also use the term “field safety corrective action” in addition to the term “corrective action.”
“corrective action taken by a manufacturer for technical or medical reasons to prevent or reduce the risk of a serious incident in relation to a device made available on the market;”
Source: MDR Article 2(68)
Although neither the MDR nor the IVDR define the term preventive action, they do use it. But only in the phrase “corrective and preventive action.” Why this mixing of the two terms is a problem is explained later in this article.
3. Preventive action
Definition
ISO 9000 defines the term preventive action as follows:
“action to eliminate the cause of a potential nonconformity or other potential undesirable situation.”
Source: ISO 9000:2015 3.12.1
Preventive actions are aimed at avoiding future nonconformities that have not yet occurred.
Examples of preventive actions
These actions can relate the design of a device to improve its safety, e.g.:
- Selecting another material or other components
- Using a more legible font on a user interface
- Introducing an input value range check
- Restricting the intended purpose
- Changing the system architecture, e.g., introducing a watchdog
Other actions might relate to quality management, e.g.:
- Ensuring better qualification of employees
- Improving a process, such as the development process
- Introducing additional code reviews
- Revising a checklist for reviewing software requirements
- Introduction of a new metric for static code analysis
If you were to take one of these actions to prevent a nonconformity that has already occurred from occurring again in the future, these actions would not be preventive actions, they would be corrective actions. In another words:
You can’t take a preventive action if the problem has already occurred. If, after a problem has occurred, you want to make sure it doesn’t occur again, that would be a corrective action not a preventive action, even though both have the same aim: to prevent a future problem.
Because most manufacturers only react when problems occur, there are a lot of corrective actions and not many preventive actions.
4. Regulatory requirements for corrective and preventive actions
a) ISO 13485
In Section 8.5 (“Improvement”), ISO 13485 requires both corrective actions (Section 8.5.2 “Corrective action”) and preventive actions (Section 8.5.3 “Preventive action”).
Manufacturers must define processes and keep records for them and provide an explanation if they do not take any corrective or preventive actions in response to a customer complaint.
Organizations must demonstrate the effectiveness of both corrective and preventive actions. The respective standard operating procedures must, therefore, require effectiveness tests.
However, ISO 13485 weakens this requirement for preventive actions with the words “where appropriate.” If an effectiveness test is inappropriate, the organization must justify this.
b) FDA
The FDA requires corrective and preventive actions in 21 CFR part 820.100. The requirements are essentially the same as those in ISO 13485.
The FDA is mainly replacing 21 CFR part 820 with a reference to ISO 13485, making the requirements for corrective and preventive action (CAPA) completely the same.
c) MDR and IVDR
The MDR and, likewise, the IVDR establish requirements for corrective and preventive actions. These include:
- The QM system must cover these actions (Article 10)
- This system must be audited by the notified bodies
- Manufacturers must implement necessary corrective actions (Article 10)
- Distributors, importers and authorized representatives must cooperate with this process
- Manufacturers must report field safety corrective actions to the authorities
- They are also obliged to decide which corrective and preventive actions are necessary using post-market data (e.g., Article 83 et seq. or IVDR Article 78 et seq.)
- In the case of clinical investigations, sponsors must report corrective actions
d) GHTF
The GHTF has published a guidance document named Quality management system -Medical Devices – Guidance on corrective action and preventive action and related QMS processes, which is worth reading. The document refers to ISO 9000 for the definitions but to the 2005 edition.
Some of the suggestions on how to implement the requirements of ISO 13485 (e.g., on root cause analysis) can also be found in the Practical Guide to ISO 13485. Auditors use both documents.
The GHTF Guidance is free of charge; the ISO Guide costs approx. 100 EUR.
5. The CAPA problem
The term CAPA stands for “Corrective And Preventive Action.” However, this combining of the two types of action is problematic for several reasons.
a) Problems with standard operating procedure
Some companies create a Standard Operating Procedure (SOP) with the title “CAPA”, in which they (only) define one common procedure for both corrective actions and preventive actions. Sometimes, they even require a preventive action for each corrective action.
In doing so, they pursue the idea that it must be ensured (“prevented”) that the problem does not occur again in the same or similar manner. But this form of “prevention” is not a preventive but a corrective action.
There can also not be only one procedure because the types of measures differ regarding inputs, roles, or regulatory requirements.
Different inputs
The employee suggestion scheme, the list of future standards and laws, technological trends, and key performance indicators point to possible future non-conformities. However, they are not yet to be understood as information that points to already existing non-conformities and whose causes the manufacturer would therefore have to eliminate as corrective action.
This means, for example, that the employee suggestion scheme is part of the process with the preventive actions but not the subject of the process with the corrective actions.
Different activities and roles
A corrective action requires different or additional activities and, in some cases, roles than a preventive action:
- Root cause analysis for corrective action may differ from the one for preventive action: In a corrective action, it is known that there is a nonconformity. Therefore, it is also sure that at least one cause exists for it. In a preventive action, it is necessary to look for the causes of a potential nonconformity.
- A decision on whether the authorities have to be notified usually only has to be taken for corrective actions.
There are many methods for finding causes of errors that have already occurred, e.g., the 5-Why method. These methods can usually also be applied to find the causes of errors that have not yet occurred: “How could XY happen?”
Different regulatory requirements
ISO 13485 has very precise requirements for handling nonconformities. This means that manufacturers have less freedom when it comes to corrections and corrective actions than they do with preventive actions.
If the MDR and IVDR had adopted the definitions contained in ISO 13485, we wouldn’t need to consider whether corrective actions as defined by the MDR and IVDR are the same as corrective actions and preventive actions combined as defined by ISO 13485.
b) Problem with “non-significant changes”
The MDR grants transitional periods for “non-significant changes.” However, according to the MDCG, what is considered a non-significant change depends on whether it is related to a corrective action.
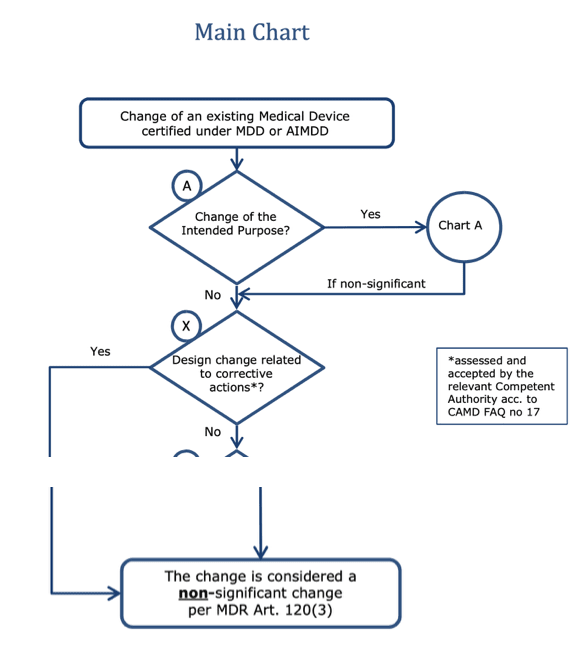
Note: Between the branch leading to “no” regarding the question “Design change related to corrective actions*?” and the box below, there are further case distinctions in the original, which are not shown here.
Do preventive actions now also have to be considered “non-significant design changes”? This would open a whole range of possibilities for manufacturers. Or does the MDR now make a precise distinction between corrective and preventive actions?
Precise definitions of terms and consistent use of these terms would prevent such discussions.
6. Conclusion
The clear separation of correction, corrective, and preventive actions makes sense and should be strictly followed by manufacturers. Standard operating procedures must regulate all these activities. Combining the processes/procedures for corrective and preventive actions as “CAPA” is inappropriate.
The fact that, of all things, the EU regulations MDR and IVDR destroy this conceptual integrity is annoying.
The Johner Institute helps you establish lean MDR-, IVDR-, and FDA-compliant QM systems that pass audits and inspections.
Change history:
- 2024-06-26: Note boxes at the beginning of the article and in chapter 4.a) added and conclusion revised
- 2023-10-06: Examples added, hint boxes added
- 2023-05-31: Editorial changes
- 2021-02-09: Article completely revised
- 2021-02-10: Link to IMDRF document and Practical Guide to ISO 13485 added