The Medical Device Single Audit Program (MDSAP) was initiated to satisfy a wish of many medical device manufacturers: To replace the various audits and inspections by authorities from different countries with one audit.
Participating in the MDSAP shall suffice for verifying effectiveness and conformity of QM systems (e.g., with ISO 13485 or 21 CFR part 820).
In this article, you will learn if this wish has really come true and at what price.
1. MDSAP: An Introduction
a) Objectives of the MDSAP
The International Medical Device Regulators Forum IMDRF, a group of international authorities and legislators, has realized that the multitude of audits and inspections of QM systems required in different target markets are suboptimal in many ways.
It is/was hoped that standardization through the MDSAP would bring many advantages.
Avoid QM overhead at the level of manufacturers and accelerate market access
Manufacturers must endure many, often redundant audits and inspections. This results in QM overhead and means efforts, costs, and time delay of marketing. MDSAP aims at minimizing these efforts.
Minimize redundant efforts and standardize interpretation of requirements
The efforts also affect international regulators: they have to formulate almost identical requirements for QM systems and continuously develop them further, as has recently been the case with the MDR. The regulators also strive for an even more uniform interpretation of the QM requirements (such as ISO 13485).
Standardize assessment
Auditors will be given assessment standards which shall ensure that different auditors preferably come to the same conclusion when assessing a case of, for instance non-conformity.
Minimize redundant efforts of auditing QM systems
Highly redundant audits and inspections do not only require a great deal of effort from manufacturers or authorities themselves, such as the FDA. These auditing efforts are to be minimized. Moreover, regulators aim at more uniform auditing procedures.
Reduce audit time
According to Health Canada, MDSAP is expected to reduce audit time by 10% for firms with up to 45 employees and by even 20% for small firms with up to 15 employees.
Make black sheep easily identifiable and increase product safety
As authorities were (and still are) often acting in isolation from one another, it is easier for black sheep among manufacturers to hide weaknesses detected by one authority from another. Hence, another of MDSAP’s purposes is information exchange, especially of audit results, between participating authorities. It is hoped for that in doing so, black sheep and unsafe products will be identified faster, thus, increasing patient safety.
Subject to standardization are audits and their documentation, whereas the MDSAP requires no standardization of manufacturers’ documentation.
b) Participants
The following countries participated in the first phase (pilot) of the Medical Device Single Audit Program:
| Country | Authority | QM requirements |
| Australia | Therapeutic Goods Administration TGA | Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3) |
| Brazil | Agência Nacional de Vigilância Sanitária (ANVISA) | Brazilian Good Manufacturing Practices (RDC ANVISA 16/2013) |
| Canada | Health Canada | ISO 13485:2016 |
| Japan | Ministry of Health, Labour and Welfare (MHLW), and Pharmaceuticals and Medical Devices Agency (PMDA) | Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No. 169) |
| USA | Food and Drug Administration FDA | QSR – 21 CFR Part 820 |
The EU and WHO merely participate as observers.
The FDA considers the pilot phase, which ended in 2017, a success:
“Based on its evaluation of the MDSAP Final Pilot Report, the MDSAP Regulatory Authority Council determined that the MDSAP Pilot had satisfactorily demonstrated the viability of the Medical Device Single Audit Program. [..]FDA will continue to accept MDSAP audit reports as a substitute for routine Agency inspections.”
The FDA largely adopts the requirements of ISO 13485, as you can read in the article on 21 CFR part 820.
c) Recognition
So far, the five countries mentioned above participate in the Medical Device Single Audit Program.

Even though those countries undertake to accept MDSAP audit results, there are minor differences, limitations, and exceptions:
| Country | Recognition |
| Australia | Yes, except for medical devices incorporating a medicinal substance or a material of human or animal origin |
| Brazil | Yes, even as initial audit; no recognition if, for example, a prior ANVISA audit has found significant deviations |
| Canada | Yes. As of 2019, Health Canada will only accept MDSAP audits. |
| Japan | Yes, except for, e.g., medical devices incorporating a material of human or animal origin |
| USA | Yes, however only substitution for routine inspections (also initial inspections), not for special inspections, e.g., “for cause inspections” |
Conclusion: Canada is the only country consequently operating the procedure model. Since 2019, Canada only accepts QM systems audited under MDSAP.
d) Auditing Organizations
Audits are conducted by so-called “Auditing Organizations“ (AO). Among them are some German notified bodies such as TÜV Süd, TÜV Rheinland, and DQS-med. Notified bodies are, however, not automatically authorized to perform audits as AO.
The FDA maintains a list of Auditing Organizations and a list of AOs recognized by ANVISA online.
2. MDSAP audits
a) Requirements and exclusions
The requirements catalog is strongly based on ISO 13485:2016. In addition, requirements of participating countries not covered by ISO 13485:2016 are incorporated. Similar to ISO 13485:2016, the MDSAP’s requirements catalog is process-oriented and audits are conducted in process groups.
There are four primary processes or process groups and three supporting processes:
- Primary Processes
- Management
- Measurement, Analysis, and Improvement
- Design and Development
- Production and Service Controls
- Support Processes
- Purchasing
- Device Marketing Authorization and Facility Registration
- Medical Device Adverse Events and Advisory Notices Reporting
If an organization does not perform certain processes, e.g., development, the respective processes do not have to be audited. A manufacturer outsourcing processes does not benefit from this simplification.
Manufacturers may exclude requirements of a certain target market if he does not sell any medical devices on that target market.
b) Tasks of the auditors
An audit under MDSAP considers interrelationships of processes. The output of a development process, for instance, is the production process’s input. The audits must be carried out according to these sequences. They must be risk-based and cover the entire product life cycle (up to decommissioning).
For each of the aforementioned process groups, the MDSAP audit model describes the following:
| Element | Example (excerpts) |
| Process name | Management process |
| Purpose of auditing this process | To verify that appropriate resources for development, production […] are provided and that […] |
| Expected outcomes (= requirements) | Commitment to provide appropriate personnel and resources for infrastructure to the quality management […] |
| Links to other processes dependent upon this process | Measurement, Analysis, and Improvement; Development; Purchasing; Device Marketing Authorization; […] |
| Auditor’s tasks | Check the manufacturer’s organizational structure and related documents to verify that they include provisions for responsibilities, personnel, resources [….] |
| References to respective subchapters, articles, and paragraphs of ISO 13485:2016 and other requirements such as 21 CFR part 820 and RDC ANVISA 16/2016 for each task | SO 13485:2016: 5.1, 5.5.1, 5.5.2, 6.1, 6.2; TG(MD)R Sch3 P1 1.4(5)(b); […] |
| References to country-specific requirements for each task | none |
| Additional information for auditors (to be found in the Companion Document) | One method for confirming that adequate resources are made available is to ask the management representative to provide several examples of recent requests for different types of resources and describe the outcomes of these requests. |
c) Frequency of audits
MDSAP audits follow the same triannual frequency as audits under 93/42-EWG or ISO 13485.
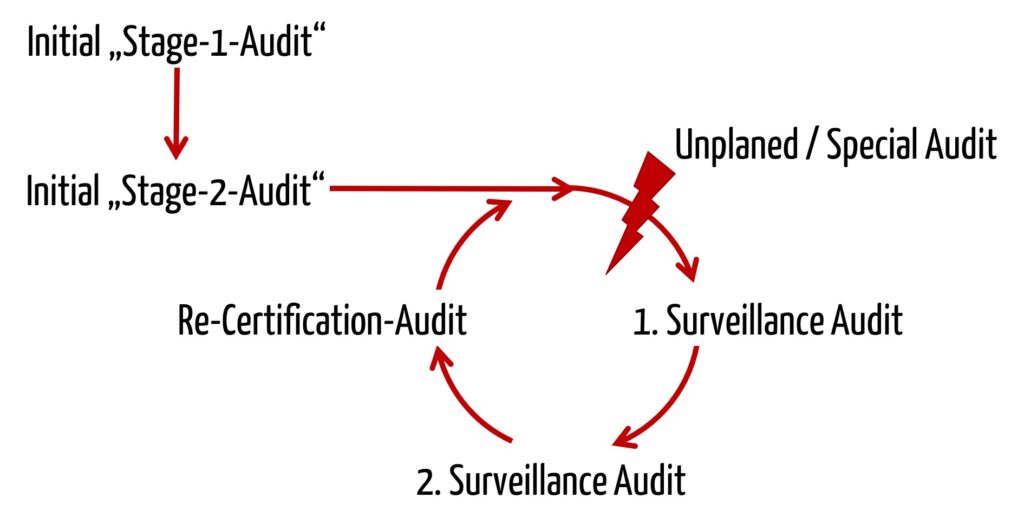
In this sequence, the MDSAP is strongly based on ISO 17021:2015, which also forms the basis for the audits according to ISO 13485. The extent to which the MDSAP is based on ISO 13485 can also be found in the description of the audit model:
“The purpose of a Stage 2 audit is to determine if all applicable requirements of ISO 13485:2016 and the relevant regulatory requirements from participating regulatory authorities have been implemented.”
d) Audit duration
The duration of MDSAP audits is calculated based on the number of tasks. This number, in turn, depends on the exclusions. For instance, it may be that a manufacturer must meet no requirements for sterile products.
In the worst case, the auditors have to complete 90 tasks, for which they are allowed between 12 and 35 minutes, depending on the type of task. 28.8 minutes are allowed for the “management” process group tasks.
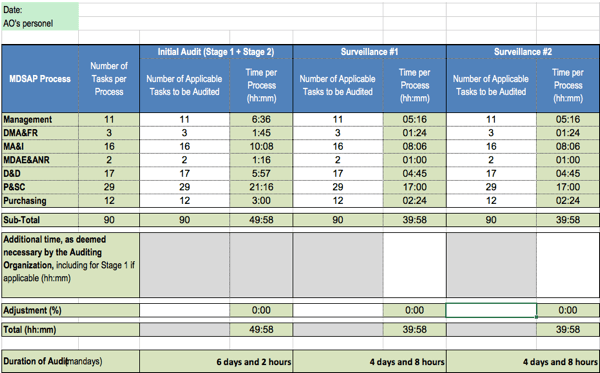
Efforts increase for auditing country-specific requirements.
e) Audit outcomes
Templates for documenting the planning, conduction, and evaluation of audits are at your disposal. Thereby, audit and non-conformance reports are standardized and can be evaluated automatically.
MDSAP sets deadlines by which Auditing Organizations must create and distribute the reports. These deadlines are more ambitious than we are accustomed to with many notified bodies.
3. Updates
- March 2018: The FDA is considering recognizing or requiring ISO 13485 as a QM standard instead of 21 CFR part 820. Although the corresponding reference has disappeared from the official website, other sources, such as the AAMI, continue to mention this. This would greatly relativize the need for an MDSAP.
- April 13, 2018: Health Canada announces on its website that it will not immediately and strictly enforce the MDSAP requirements.
- February 21, 2021: The FDA revises the list of procedures and forms on its website.
- April 1, 2023: The MDSAP AU P0002.008 is published and can also be found on the FDA website.
- August 6, 2024: The MDSAP AU P0002.009 is published and can also be found on the FDA website.
4. Conclusion
For manufacturers, a major advantage of the MDSAP is that it can reduce the number of audits and inspections by the authorities of the various target markets. Nevertheless, manufacturers were hesitant during the MDSAP pilot phase. This may be due to the following disadvantages and challenges:
- Transparency through exchange of information between all participating countries may result in other countries’ reacting to negative audits with additional inspections. The FDA, for instance, explicitly reserves the right to impose additional inspections.
- The list of requirements tends to increase as it combines requirements of various countries.
- The internal quality management team must increasingly deal with international requirements.
- One of the most important target markets got left out: Europe.
Anyone who wants to bring medical devices into the Canadian market will have no choice: The Medical Device Single Audit Program will be the only possibility. Participation means weighing the pros and cons for all other manufacturers, at least as long as Europe does not abandon its observer role and recognize the MDSAP.
The switch to MDSAP is time-consuming. So it’s best to start in good time.
The relatively transparent and standardized regulations on activities, documentation, and evaluation should help to ensure that audits are carried out more uniformly worldwide.
However, the promised alignment of efforts with the risk posed by a manufacturer’s medical devices and the manufacturer’s size still leaves much to be desired. This makes the MDSAP a rather higher hurdle than ISO 13485 audits, especially for small manufacturers with less critical devices.
The Johner Institute supports medical device manufacturers and their service providers in establishing lean QM systems and preparing for audits and inspections.
Change history:
- 2024-10-24: Reference to update of the MDSAP AU P0002.009 added
- 2024-05-17: Reference to update of the MDSAP AU P0002.008 added
- 2022-03: Corrected content error in chapter 1.c) (FDA also accepts MDSAP for initial inspection, see comments)
- 2021-03: Chapter “Updates” introduced
