Software as medical device (SaMD) refers to (independent) standalone software that is a medical device but not part of one.
It should not be confused with medical device software as defined by the EU.
As a manufacturer, when do you have to qualify software as medical device, and when as medical device software? Find out here – and use this knowledge to avoid legal consequences and unnecessary expense.
The decision as to whether software is a medical device is referred to as qualification. Colloquially, the term classification is often used. Classification is, however, understood to mean “risk classification.” This is the division into classes that define the regulations to determine the approval procedures. For example, the MDR recognizes classes I, IIa, IIb, and III.
A) Definitions
1. Software in the medical context
A distinction is made between software in the medical device environment:
- Software as part of a medical device, e.g., as embedded software of a medical device
- Software as an independent medical device (standalone software)
- Software that is an accessory for a medical device
- (Standalone) software that is not a medical device software
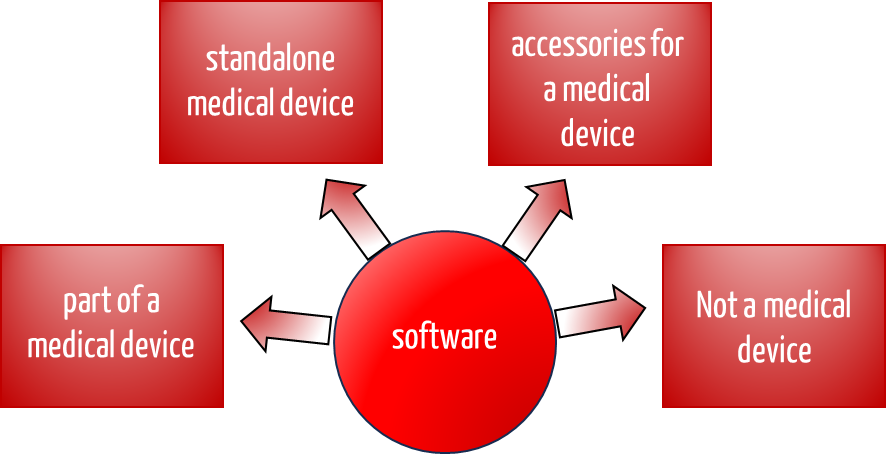
Depending on this classification, you as a manufacturer must comply with different regulations. This article will help you qualify your software correctly.
2. Definitions
There are several definitions of what is meant by software as medical device. One definition comes from the IMDRF, the International Medical Device Regulators Forum.
“software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device“
Source: IMDRF/SaMD WG/N12FINAL:2014
According to the German Federal Ministry of Health, the following definition of the term “medical software” is planned in the MDR guidelines, as developed by the MDCG:
“Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.”
In addition to the definition, the MDCG writes:
Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.
Source: MDCG 2019-11
This means that medical device software includes both independent software (i.e., standalone software) and control software (often also referred to as functional software).
This definition and the MDCG’s guidance document strongly impact the classification of software and the applicability of Rule 11.
Read more about the MDCG and Rule 11 here.
The IMDRF provides further key definitions in the SaMD context in the document IMDRF/SaMD WG/N10Final:2013.
B) Qualification (classification) of software as medical device
The decision as to whether software counts as a medical device is referred to as qualification, even if the term classification is often used colloquially.
The intended purpose of the manufacturer decides …
The question of when software qualifies as a medical device only arises in the case of standalone software. The short answer is that software is a medical device if the manufacturer has intended it to diagnose, treat, or monitor diseases and injuries.
More precisely and formally, software is a medical device if the manufacturer’s intended purpose meets the definition of the term “medical device” in Article 2(1) of the MDR.
… and not primarily the software functions
The decisive qualification factor is the manufacturer’s intended purpose rather than the software’s function. In the case of software that records vital signs, there are two ways of arguing this, which lead to different assessments:
- The manufacturer says: The recording is for documentation purposes only. Then, the device is not a medical device.
- The manufacturer says: The doctor can use these vital signs (and possibly their graphical representation) to recognize trends and thus select the right medication. Then, the identical device is a medical device.
And what if there is no intended purpose?
If the manufacturer does not formulate the intended purpose clearly and without contradiction (manual, packaging, website, marketing materials, etc.) and does not classify its device as a medical device, the “dear competition” may send a warning letter. If this is the case for you, then contact us. We are familiar with this.
The judge will probably appoint an expert if the matter ends in court. This expert will, in turn, try to deduce the use intended by the manufacturer (intended purpose) from the documents (manual, etc.) and the functions(!). It is doubtful whether the expert’s conclusions will be in your favor.
Who decides on the qualification/classification of software as medical device
Despite the definition of the term, manufacturers and authorities often feel uncertain about whether software should be classified as a medical device. This is one of the reasons why many decision aids have been published, including the ones presented in this article.
The manufacturer makes the decision as to whether software is to be classified as a medical device. Notified bodies can issue expert opinions. However, this does not provide the manufacturer with legally binding information. Inquiries to the BfArM are generally not answered promptly. Ultimately, and this may sound cynical, only a judge will finally answer the classification question in case of a lawsuit.
If you are unsure whether your software is a medical device, then contact
- the Federal Institute for Drugs and Medical Devices BfArM (the response time is not always quick, however)
- notified bodies (we can provide you with contacts here)
- the Johner Institute (we constantly answer such questions and prepare expert reports)
Are you unsure whether your medical software is a medical device? Do you need an expert opinion? We are happy to help.
C) Decision-making aids for the qualification/classification of software as medical device
The topic of “classification of software as a medical device” not only concerns medical device manufacturers but also authorities, committees, and associations. These have published a series of documents that are intended to serve as decision-making aids. We present the following documents:
- COICR Contribution
- EU: Manual on Borderline and Classification
- UK Medicines and Healthcare Products Regulatory Agency
- International Medical Device Regulator Forum IMDRF
- MEDDEV 2.1.6
- Swedish authorities
- Asian Harmonization Working Group
- SwissMedic
- British MHRA
- FDA
- Health Canada
- MDCG (particularly relevant in Europe)
- Australian Ministry of Health
1. COICR Contribution
The “European Coordination Committee of the Radiological, Electromedical and Healthcare IT Industry” has published a “Decision Diagram for Qualification of Software as Medical Device.”
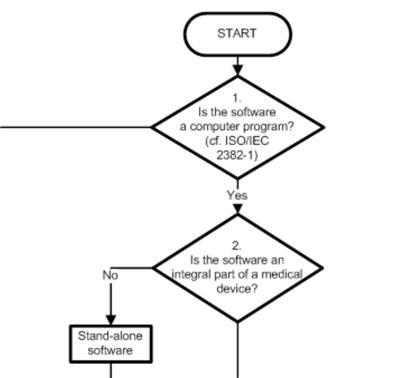
The value of this document is debatable. On the one hand, it does not contain anything new (ultimately, only the legal definition counts). On the other hand, a lot of work has been done here to support manufacturers and developers.
2. EU: Manual on Borderline and Classification
The manual (V 1.22 (05-2019)) originates from the EU and uses examples to distinguish medical devices from non-medical devices and provide help with qualification and classification. Some of the examples also refer to software:
- Picture archiving and communication systems
- Mobile application for processing ECGs
- Mobile application for the communication between patients and caregivers while giving birth
- Mobile medical application for viewing the anatomy of the human body
The EU has published an infographic to help you decide whether software counts as medical device software (not SaMD!).
3. Medicines and Healthcare Products Regulatory Agency
The British Medicines and Healthcare Products Regulatory Agency has published a document entitled “Borderlines with medical devices.” Please read Chapter 9, the sentence on “Telecare Alarm Systems.” I would be interested in your assessment.
4. IMDRF
This document from the International Medical Device Regulator Forum IMDRF is one of the most relevant. It defines the following terms:
- Software as Medical Device (SaMD)
- Medical Device
- In Vitro Diagnostic Medical Device
- SaMD Changes
- SaMD Manufacturer
- Intended Use/Intended Purpose
With ten pages, the document is pleasantly short and worth a look. The references are interesting, including the sources:
- GHTF/SG1/N55:2008 Definition of the Terms Manufacturer, Authorized Representative,
- Distributor and Importer
- GHTF/SG1/N70:2011 Label and Instructions for Use for Medical Devices
- GHTF/SG1/N71:2012 Definition of Terms Medical Device and In Vitro Diagnostic
- Medical Device
- ISO/IEC 14764:2006 Software Engineering – Software Life Cycle Processes – Maintenance
- Maintenance
The IMDRF returns to the definition of the term medical device (software as medical device) and lists cases in which standalone software does not count as such. Conversely, the document provides information on when this software meets the definition, e.g.,
- mitigation of a disease,
- provide information for determining compatibility, detecting, diagnosing, monitoring
- or treating physiological conditions, states of health, illnesses, or congenital deformities,
- aid in diagnosis, screening, monitoring, predisposition, prognosis, prediction, determination of physiological status,
- aids for persons with disabilities.
Whether these are groundbreaking findings for classifying software as a medical device is certainly debatable. After all, the classification assistance does not go much further than the definition of a medical device.
5. MEDDEV 2.1.6
Note that the MDCG document 2019-11 discussed below makes MEDDEV 2.1/6 obsolete for software regulated by the MDR!
MEDDEV 2.1/6 published an updated version of the Guidance on the Qualification and Classification of Stand Alone Software used in Healthcare within the Regulatory Framework of Medical Devices in July 2016. Software was divided into the following categories:
- Software that is not a medical device, such as development tools or documentation systems, which include HIS (but only as long as they are not used for diagnosis or therapy)
- Software as a component of a medical device, i.e., device software
- Software that is itself a medical device
- Software that is an accessory
A decision tree that has been revised compared to the 2012 version was intended to help determine the correct class:
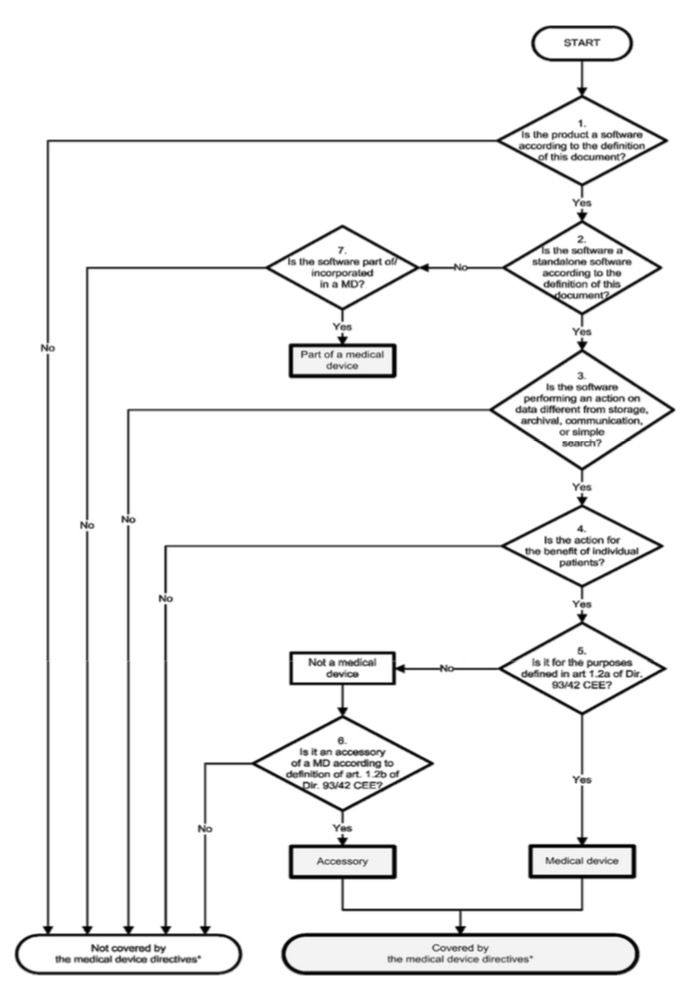
This document was one of the most important in this context.
This EU guideline also assisted in the classification of IVD software.
Read more about the classification of IVD software here.
6. Swedish authorities
The Swedish authorities issued the guideline “Medical Information Systems – guidance for qualification and classification of standalone software with a medical purpose.” The link is no longer available since February 2020.
7. Asian Harmonization Working Group – Software Qualification and Classification
The document of the Asian Harmonization Working Group repeats the definitions in the context of “Software as Medical Device,” such as standalone software, “Mobile Medical Application,” and “Health Software.” It distinguishes between three types:
- Software that is part of a medical device software (often referred to as embedded software)
- (Standalone) software that is an accessory for a medical device, e.g., for forwarding data
- Standalone software that is itself a medical device (software as medical device SaMD) and is made available on a data carrier via download or web-based
The document also presents known considerations for classifying software as a medical device and essentially discusses the sources already mentioned above:
- The IMDRF publication (see above)
- The Australian Therapeutic Goods Association (TGA) references European and US publications. It does not classify documentation software as a medical device, nor does it classify software that only performs simple calculations that are not based on patient-specific data. If the software controls or influences another medical device, it falls into the same class as the medical device.
- The Chinese Food and Drug Administration (CFDA) has published a detailed guideline for registering software as a medical device, which uses the safety classification according to IEC 62304.
- Of the documents published in Europe, MEDDEV 2.1/6 (see above) is cited, along with tabular examples of software that could be classified as a medical device or a non-medical device. The classification rules of Annex IX of the MDD are also cited.
- The document briefly discusses the requirements of Health Canada and mentions, in particular, an FAQ document that provides examples and rules for classifying software as a medical device. A table gives specific examples.
- The guidelines of the Japanese MHLW (Ministry of Health, Labor and Welfare) state that standalone software can be a medical device and that further recommendations are currently being developed.
- An overview of the FDA’s requirements (see above) concludes the series of legal areas examined.
The document summarizes similarities and differences in tabular form in the summary. It also discusses software used for data communication. This software is not classified as a medical device in Europe, but it is in most other areas of law.
8. SwissMedic
SwissMedic has also commented on the topic in the AW leaflet Standalone medical device software. It summarizes many of the sources mentioned above without providing any new findings. What is interesting is the clear statement that the same requirements must be met for software produced “in-house” (e.g., within hospitals). The Swiss authority even writes:
“By definition, software produced in-house is not placed on the market, but its use by a healthcare professional is considered equivalent to placing it on the market for the first time.”
9. The British MHRA
In September 2022, the UK Medical & Healthcare products Regulatory Agency (MHRA) published a document to explain when software is a medical device and when it is not.
The rules are essentially the same as in the MDCG 2019-11 guideline.
The MHRA has gone to great lengths to formulate the requirements and rules in a visually appealing and easy-to-understand way. Unfortunately, it also confuses intended purpose (e.g., “Prevention of Disease”), functions (“Stores or transmits medical data”) and technical characterizations (e.g., “Database without internal language”), which harms clarity.
10. FDA for the qualification and classification of software as medical device software
a) Food, Drug, and Cosmetic Act (FD&C)
In summer 2017, the US Food, Drug and Cosmetic Act, or FD&C for short, revised the definition of the term “medical device” specifically for software. However, this description has become so cryptic that the FDA published a guidance document in December 2017 to set out its law interpretation. While this outline only applies to decision support systems, it is highly applicable to other standalone software.
Read more about the FDA Guidance Document on Decision Support Systems.
The AAMI document on the agile development of medical device software is helpful.
In September 2019, the FDA published the guidance document Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act. In it, the authority once again sets out its view of things in detail, gives examples, and refers to other documents (some of which are mentioned below).
b) General Wellness: Policy for Low Risk Devices
The Food, Drug & Cosmetic Act was revised at the end of November 2016. It no longer(!) defines software “for maintaining or encouraging a healthy lifestyle and is unrelated to the diagnosis, cure, mitigation, prevention, or treatment of a disease or condition” as a medical device.
In its guidance document General Wellness: Policy for Low-Risk Devices, the FDA confirms that it does not wish to investigate whether such “low-risk general wellness products” are medical devices, nor whether such devices classified as medical devices meet the regulatory requirements.
However, the prerequisites for this are
- The devices are intended solely for general wellness (“wellness”) as defined in this document.
- The devices represent a low risk.
Manufacturers must not make any reference to diseases that the devices are intended to diagnose or treat. Instead, it is about, for example
- weight management (but not treatment of obesity!),
- physical fitness,
- stress management,
- sleep.
However, the FDA also allows devices related to specific diseases in this category if they may help
- minimize the risks of certain chronic diseases,
- live better with certain chronic diseases.
The guidance document gives examples and provides a small decision tree.
Conclusion: With this document, the FDA clarifies when devices – including software – are excluded from FDA monitoring. Europe would also like to see this. The authority also excludes many more devices from monitoring than European authorities and regulators.
c) Policy for Device Software Functions and Mobile Medical Applications
The 2016 guidance on mobile medical apps already delimited apps that are
- are not a medical device,
- are a medical device but are not subject to FDA review,
- are a medical device subject to FDA review.
With the 2019 update, the FDA has also extended the scope to other software and now often refers to “Software Functions.” Mobile apps are now only one type of software that has such functions.
This means that you should also consult the document if you need to decide whether software that is not a mobile medical app is a medical device.
You can read more about this in the article on mobile medical apps.
d) FDASIA Report
The FDA is a co-publisher of the FDASIA Report. FDASIA stands for §Food and Drug Adminstration Safety and Innovation Act§, which requires the FDA to produce a report,
“that contains a proposed strategy and recommendations on an appropriate, risk-based regulatory framework pertaining to health information technology, including mobile medical applications, that promotes innovation, protects patient safety, and avoids regulatory duplication.“
In this report, the FDA again distinguishes (as in the guidance document on mobile medical apps):
- Non-medical devices
- Medical devices for which the FDA requires compliance with legal requirements (e.g., approval procedures, Quality Systems Regulations)
- Medical devices for which the FDA does not do so
This tripartite division is surprising. So, laws do not always have to be complied with? The FDA justifies this differentiation with a “risk-based approach”:
“In applying a risk-based approach, FDA does not intend to focus its regulatory oversight on these products/functionalities, even if they meet the statutory definition of a medical device.“
It then gives specific examples where it considers these exceptions to be justified:
- Evidence-based clinician order sets tailored for a particular condition, disease, or clinician preference;
- Drug-drug interaction and drug-allergy contraindication alerts to avert adverse drug events;
- Most drug dosing calculations;
- Drug formulary guidelines;
- Reminders for preventative care (e.g. mammography, colonoscopy, immunizations, etc.);
- Facilitation of access to treatment guidelines and other reference material that can provide information relevant to particular patients;
- Calculation of prediction rules and severity of illness assessments (e.g., APACHE score, AHRQ Pneumonia Severity Index, Charlson Index);
- Duplicate testing alerts;
- Suggestions for possible diagnoses based on patient-specific information retrieved from a patient’s EHR.
It is not entirely clear what statistical analysis the assumptions that the product mentioned above classes pose few risks are based on. Such clinical decision support systems are often treacherous because the risks are less obvious than those of classic medical devices such as ventilators.
Due to the amendment to Section 520 of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 360j) mentioned in the next section, the FDA must rewrite some guidance documents, in particular those on Medical Device Data Systems MDDS and on Mobile Medical Apps.
A new guidance document gives specific examples of when software is no longer to be classified as a medical device:
- Software for administrative support
- Laboratory information systems (as long as they only provide administrative support, storage, transport, and conversion of laboratory data)
- Software that aims to support a healthy lifestyle, e.g., an app that records and displays exercise or an app that warns the user if they eat too much or eat unhealthily. Diaries and social games are also among the examples.
- Electronic patient records, but only if three conditions are met:
- They are maintained by or under the supervision of healthcare professionals.
- They are not subject to the Health IT Certification Program.
- They are not used for the analysis of records, including images, for the purpose of diagnosis, treatment, palliation, or monitoring.
- Software used only for the transmission, storage, format conversion, and display of laboratory data and other device data, as long as no analysis of this data is performed. This means that Medical Device Data Systems are no longer medical devices. PACS also no longer count as medical devices.
Accordingly, the FDA must revise the above-mentioned guidance documents.
e) Digital Health Policy Navigator
The FDA provides support in the qualification and classification of device software functions. For this purpose, it has developed a Digital Health Policy Navigator that helps with decision-making in seven steps.
You can learn more in this FDA Draft Guidance on “Existing Medical Software Policies.”
11. Health Canada
Health Canada has published the draft “Guidance Document – Software as a Medical Device (SaMD).” It is essentially based on the IMDRF “Framework” mentioned above and adopts its definitions. It supplements its ideas with examples.
a) Medical purpose
Health Canada gives examples of intended medical purposes:
- Acquiring, processing, or analyzing medical images
- Acquiring, processing, or analyzing information obtained from an IVD
- Acquiring, processing, or analyzing information, measurements, or signals from a monitoring or imaging device
- Assisting or providing recommendations to patients or healthcare professionals for the prevention, diagnosis, treatment, and mitigation of disease or injury
However, Health Canada does not qualify all decision support systems as medical devices.
b) Examples of software that is not a medical device
Health Canada also helps with examples of software that the authority does not consider to be medical devices:
- Pure communication systems such as MDDS, telephony, etc.
- Software with purely administrative purposes, including workflow, patient management, scheduling
- Software that contributes to a healthier lifestyle, such as wellness apps
- Electronic health records
- Software that provides access to literature data
- Software that warns of drug interactions
- Software that does not trigger immediate action
- Software for simple medical calculations
c) Risk classification
Health Canada also bases its risk classification on the IMDRF document but allows a lower classification in some places.
12. MDCG
a) Overview
The document MDCG 2019-11 regulates the qualification and classification of medical device software under the MDR. It replaces the above-mentioned MEDDEV 2.1/6. The classification scheme is central for many manufacturers:
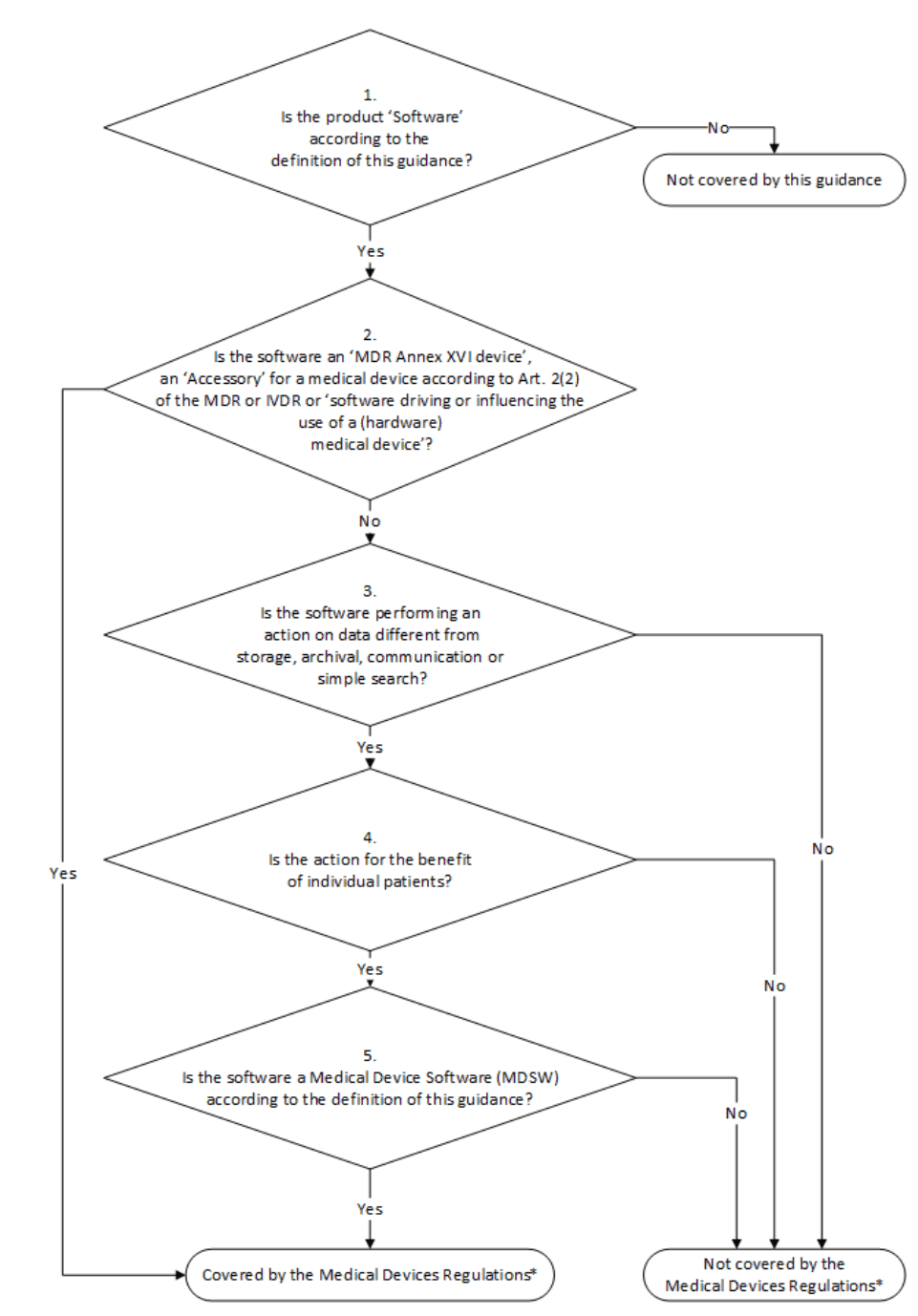
The last case distinction, “Is the Software a Medical Device Software according to the definition of this Guidance?” is particularly relevant.
As a reminder:
Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.
MDCG 2019-11
The MDCG points out that it is irrelevant for the classification as MDSW,
- where the software is running (in the cloud, on smartphone, on the server),
- who uses the software (lay persons, healthcare professionals),
- whether the software controls a medical device or not, and
- whether the software runs independently or is part of a medical device.
b) Examples
The MDCG gives examples of such medical device software (MDSW):
- Standalone software used for diagnosis or treatment, including software to assess a child’s risk of trisomy 21 based on the mother’s serum markers
- Software that calculates the risk of prostate cancer from a patient’s ultrasound images and laboratory parameters
- Software for a mass spectrometer that uses data to detect microorganisms and their resistance to antibiotics
- An app that sends alerts to the patient or doctor if it detects an arrhythmia from irregular heartbeats
- Software that measures glucose values, uses them to determine the insulin dose, and uses them to control an insulin pump (possibly even as a closed-loop system)
- Cloud-based software that performs a point-of-care test
- Provision of expert assistance for clinical decisions (e.g., planning of radiation therapy doses)
The MDCG also gives examples of “non-MDSW”:
- Collecting and managing administrative patient data
- Storing patients’ medical history
- Performing invoicing and other billing functions
- Linking to the social security system for reimbursement
- Linking to systems for prescribing and dispensing medicines
Also, please read our article on when software counts as an IVD. This qualification and classification also follows from the MDCG document. It is also important to assess when software falls into class I according to MDR.
c) Special case: Software only for controlling hardware
If software is used exclusively to control the hardware of a medical device and has no medical purpose of its own, it is only to be regarded as an accessory for a medical device. An additional classification in accordance with Rule 11 is then out of the question.
d) Criticism of the MDCG document
Criticism arose just a few days after publication. Professor Antonio Bartolozzi published a presentation on LinkedIn:
The Johner Institute assesses this criticism as follows:
- New and unagreed definition
The new definition of medical device software (MDSW) indeed differs from existing definitions. This can cause confusion. At the same time, this separate definition represents the actual concept, which can and should(!) lead, among other things, to software that is part of a physical medical device, classifying the entire medical device higher. - New concept
Further points of criticism in the presentation appear to be due to the fact that the author does not want to come to terms with this concept or does not understand it. However, the MDCG document would have benefited from a deliberate differentiation and comparison of the concepts. - Binding nature of the MDCG documents
It is legitimate to question the legally binding nature of the MDCG documents. Rules are laid down here, bypassing parliamentary processes or other forms of consensus building (as in standardization), which are de facto interpreted as binding. Even ECJ judges have relied on such documents. - Examples
Prof. Bartolozzi also strongly criticizes the examples. Of course, hospital information systems are used to obtain information for diagnosis and treatment. However, this is not in their intended purpose. It is true that these systems are therefore misclassified. However, it was not the MDCG’s task to regulate this.
One can also agree that a PDMS is a medical device. Here, the MDCG is somewhat unhappily wriggling out of the matter by referring to modules. - Inconsistencies
In fact, the classification of medical devices with identical intended purposes should not depend on whether they are software or hardware products.
Conclusion: The criticism, which in some places appears somewhat heavy-handed with countless exclamation marks, cannot be entirely dismissed. Rule 11 is unsuitable in its current form. The MDCG document only partially remedies this shortcoming. This is also due to the fact that it
- introduces new definitions and concepts without sufficiently explaining and delimiting them,
- bends existing concepts (e.g., IMDRF),
- is formulated in such a way that it would not pass every usability test, and
- seems at least somewhat arbitrary or contradictory to existing rules in some places.
The Johner Institute does not believe that the classification proposed by Mr. Bartolozzi is the solution. It would mean that even more devices would fall into class III than the MDCG proposes.
He himself argues that the classification should be risk-based but then refers back to Rule 11, which only considers the severity of possible harm.
The new edition of the MDCG document from June 2025 does not change the classification in principle. However, the MDCG reinterprets “prevention” as “diagnosis”:
a device intended to prevent the risk of illnesses or pathologies by […] can be considered as a device providing information which is used to take decisions with diagnosis purpose […] and in this case is in class IIa.
The MDR clearly distinguishes between the terms “prevention,” “diagnosis,” “therapy,” “prediction,” and “prognosis.”
The additional examples of software as a medical device and software that is not considered a medical device provide a little more clarity. It is commendable that the MDCG emphasizes the intended purpose as the basis for qualification and classification, even if its own flowchart contradicts this. There, the decision on qualification is recommended based on functionality (“archive,” “storage,” “query”).
13. Australia
In the document Exemption for Certain Clinical Decision Support Software Guidance on the exemption criteria, the Australian TGA describes the circumstances under which a clinical decision support system is not subject to regulation.
This is the case if three conditions are met:
- The software is used solely to make recommendations to “health professionals” to diagnose, prevent, cure, and mitigate disease and injury.
- It does not directly analyze or process medical image data or biosignals from other medical devices (including IVDs).
- It is not intended to replace the judgment of health professionals regarding diagnosis or therapy.
Many devices benefit from these facilitations.
D) Examples of software as a medical device
1. Website as medical device
Question
Heise Online reported on a start-up whose business idea was to diagnose skin diseases. The company’s website guided users through a questionnaire and allowed them to upload photos of their own skin. You received an answer within 48 hours – all for just EUR 29. The website is now offline.
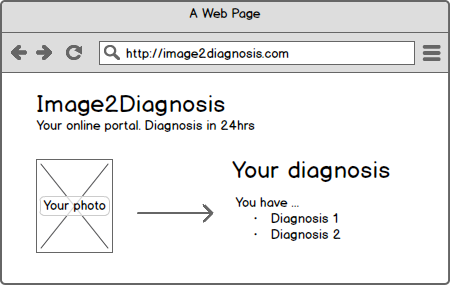
The question quickly arises whether a website is a medical device. Websites can generally be medical devices, but what about a specific website? Does it also fall under this definition?
Evaluation
2007/47 clarified that standalone software also falls under the definition of a medical device if it is intended for diagnosing, treating, and monitoring diseases and injuries, to put it in a nutshell. It stands to reason that the website in question is intended to diagnose diseases, as the service offered is the diagnosis of skin diseases.
For the classification (not only of websites) as a medical device, the question of whether something can happen is not decisive.
2. Web service as medical device
a) Example
The company Promedas offered a web service that provides an API for medical diagnoses.
The manufacturer’s website stated:
Promedas provides a clinical expertise system to medical professionals. The Promedas API can be integrated into existing medical systems that contain a patient file database. Using this data, Promedus can provide a differential diagnosis based on the data contained in a patient’s file. The API is currently in a developer beta. To access the API, contact Promedas.
b) Questions
This immediately raises questions such as the following:
- Is this web service itself already a medical device?
- If you integrate this service into your own software, how do you treat it according to the standard? As a SOUP?
- If this web service has a simple GUI, it can theoretically be accessed and benefited from worldwide. Does it then automatically have to be designed for all international legal requirements? Even if there were an intended use that the service may only be used in the EU, it would immediately become apparent during market surveillance that it is also being accessed in the USA. Would it, therefore, also have to meet FDA requirements?
c) Evaluation
Question 1: Until recently, the answer should have been “no.” The web service is not a medical device because it does not serve to diagnose or treat patients but is intended to be integrated into a system whose intended purpose is diagnosis or treatment.
According to the ECJ ruling (see below), this assessment must be revoked. Accordingly, a device’s “modules” may be classified as a medical device, even if the device into which the components are integrated is not a medical device.
There are other ways to integrate a component that is classified as a medical device into a product without “infecting” the device, i.e., making it a medical device as well.
If you would like further information, please do not hesitate to contact us.
Question 2: It is conceivable to declare a web service as a SOUP and integrate it into a medical device as a component whose development does not meet the requirements of IEC 62304. This procedure and this component would, of course, have to be evaluated as part of risk management.
Question 3: An exclusion in the intended purpose would already get you off the hook. If an authority threatens to take measures because clinics or doctors are obviously ignoring this requirement, this could be solved reasonably well via IP filtering. However, a European authority has no power in the USA and vice versa. It would, therefore, never take action in another legal area.
3. Document Management Systems
a) Question
As part of our free micro-consulting service, we were asked whether Document Management Systems (DMS) are medical devices. It was argued that errors in a DMS could result in harm to patients. On the other hand, doctors would always make the final decision.
b) Evaluation
Both considerations are important but not relevant for classification as a medical device:
Software never directly inflicts harm. It is always people or hardware that ultimately injure or harm the patient.
What is relevant, however, is the manufacturer’s statement about what its customers can and should use the device for. If the manufacturer says the system is only for documentation purposes, it is not a medical device. If, on the other hand, the manufacturer states that the system is to be used to store X-ray images for monitoring purposes, for example, the system is used for therapy and is, therefore, by definition, a medical device.
By formulating this intended purpose, the manufacturer can make his device a medical device or not. The intended purpose is not just a document. Instead, the manufacturer documents this in a variety of ways:
- In the instructions for use
- With the online help of the device
- On its website
- In marketing materials such as flyers
- At trade fairs
- Even in conversations
The Johner Institute, therefore, recommends that DMS manufacturers articulate themselves clearly. This can also include explicit exclusions of functionalities or application scenarios.
Do you have questions about the classification of your device? Then, please benefit from our free micro-consulting service.
4. Hospital Information System (HIS) as medical device
a) Question
The task of Hospital Information Systems (HIS) is to support users in diagnosing, monitoring, and treating patients. The fact that these systems occasionally (indirectly) endanger patients is also not unknown. This can be the case when they
- mix up patient data,
- lose data,
- react very slowly to user input (e.g., in the event of a malware attack),
- no longer respond at all, no longer start, fail, or
- do not forward information to other systems.
b) Evaluation
The decisive factor for whether this software is to be classified as a medical device is not the hazard but only the conformity of the intended purpose with the definition of the term medical device.
- A system that is only intended for documentation or billing purposes does not fall under this definition.
- A system that derives diagnostic or therapeutic recommendations from entered values and gives warnings in the event of drug interactions, for example, does.
c) Consequences for the operators
As no HIS manufacturer will survive in the long term if their systems can do nothing other than display data that has been entered, all HIS will become medical devices in the medium term. But nobody wants to hear that, especially not the operators, namely the hospitals.
The reason is apparent: as soon as an HIS is classified as a medical device, it is subject to the Medical Device Operator Ordinance. This requires, among other things:
- Persons who use, operate, and maintain the system must be trained. This must be documented.
- The system must be reviewed regularly, at least every two years.
- All safety-relevant functions and design features must be checked after every maintenance measure (software update).
But how are hospitals supposed to do this? How are they supposed to thoroughly check that a HIS with diagnostic functionality still works after a software update? That it still works flawlessly with the RIS in the same network segment? That there are no repercussions for ultrasound devices that are also connected to the network?
The only way to address these almost infinite combinations of possible causes of failure is to employ risk management to prioritize testing steps.
Some companies classify their systems as medical devices, such as GE Healthcare’s Centricity information system, which has received CE marking (in accordance with MDD, 93/42/EC). Here is the corresponding press release from GE (in German).
5. Software as an IVD
Software can be classified not only as a medical device (according to MDR/MDR) but also as an IVD (according to IVDD/IVDR).
Read more about IVD software here.
6. ChatGPT as medical device
You know or suspect that ChatGPT also provides answers to medical questions. One law firm, therefore, concludes:
“In our legal opinion, this software falls under regulation as a medical device in Germany and Europe.”
Pharmacy Adhoc (German)
E-HEALTH-COM also reports (German) that a “law firm specializing in digital medical devices” has “requested the regulatory classification of the AI-based software ChatGPT as a medical device” from the BfArM.
The reason stated is the realization that “ChatGPT falls within the scope of the Medical Device Regulation (MDR) due to its diagnostic and therapeutic uses.“
The Johner Institute agrees that ChatGPT can be used diagnostically and therapeutically. However, it cannot understand the conclusion that ChatGPT has become a medical device simply because it can be used this way.
The intended purpose of the manufacturer is decisive. It can be assumed that OpenAI does not intend its devices to be used for diagnostic or therapeutic purposes. In its terms of use, the company states:
“You must not use any Output relating to a person for any purpose that could have a legal or material impact on that person, such as […] medical, or other important decisions about them.”
If, on the other hand, a medical device uses ChatGPT, then the entire device must meet the regulatory requirements. Although ChatGPT could be declared as a SOUP, the specification, and verification of this component are likely to be just as difficult as proving a positive benefit-risk ratio.
It will be interesting to see whether and how the BfArM reacts. It is up to the federal states to enforce the legal requirements…
Learn all about the IEC 62304 standard and how to implement the requirements simply and practically. With over 25 videos and a complete set of an IEC 62304-compliant software file, we support you in successfully certifying your software.
7. Further examples
Many other examples can be found in Annex I of the MDCG guidance document:
- Hospital Information Systems
- Decision Support Software
- Electronic Patient Record Systems
- Clinical Information Systems – CIS/Patient Data Management Systems – PDMS
- Pre-hospital Electrocardiograph (ECG) System
- Picture Archive Communication System (PACS)
- Telemedicine systems
- Telesurgery
- Web systems for monitoring of data
- Laboratory Information Systems (LIS) and Work Area Managers (WAM)
- IVD: Expert system
- IVD: Interpretation of raw data
- IVD: Home care monitoring, wired or mobile
- IVD: Image Management System (IMS)
E) Special case: Components as medical devices
1. MEDDEV 2.1/6: Only “medical modules” must fulfill MDD
In July 2016, the EU Commission published a second version of the MEDDEV 2.1/6 guideline (see below) with the long title “Guidelines on the Qualification and Classification of Stand Alone Software used in the Healthcare within the Regulatory Framework of Medical Devices.”
This document determines that software can consist of modules. Only those modules that have a corresponding medical intended purpose must meet the requirements of the Medical Devices Directive. This is what it says:
“Some stand alone software may break down into a significant number of applications for the user where each of these applications is correlated with a module. Some of these modules have a medical purpose, some not. The modules which are subject to the medical device Directives (Figures 1 and 2) must comply with the requirements of the medical device Directives and must carry the CE marking. The non-medical device modules are not subject to the requirements for medical devices.“
The MDCG continues to use this approach. It writes:
Computer programmes used in healthcare can have applications which consist of both medical device and non-medical device modules.
MDCG 2019-11
The modules which are subject to the Medical Devices Regulations must comply with the requirements of the medical device regulations and must carry the CE marking. The non-medical device modules are not subject to the requirements for medical devices. It is the obligation of the manufacturer to identify the boundaries and the interfaces of the different modules.
An example of such software could be a Hospital Information System consisting of non-medical device modules (billing, master data management, etc.) and medical device modules (e.g., drug testing, radiological findings).
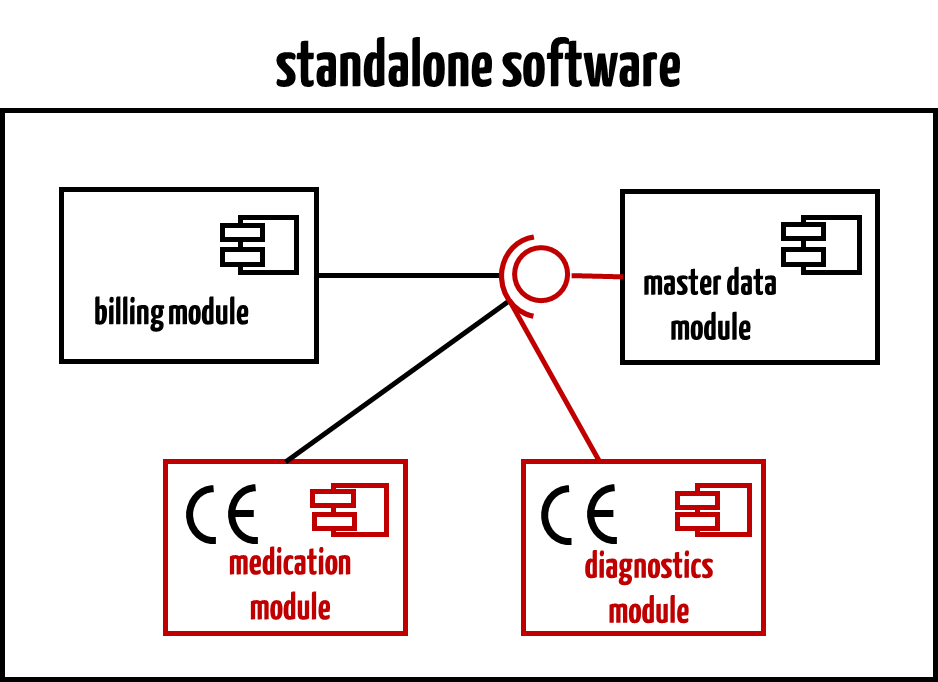
Read more about software modules and software items here.
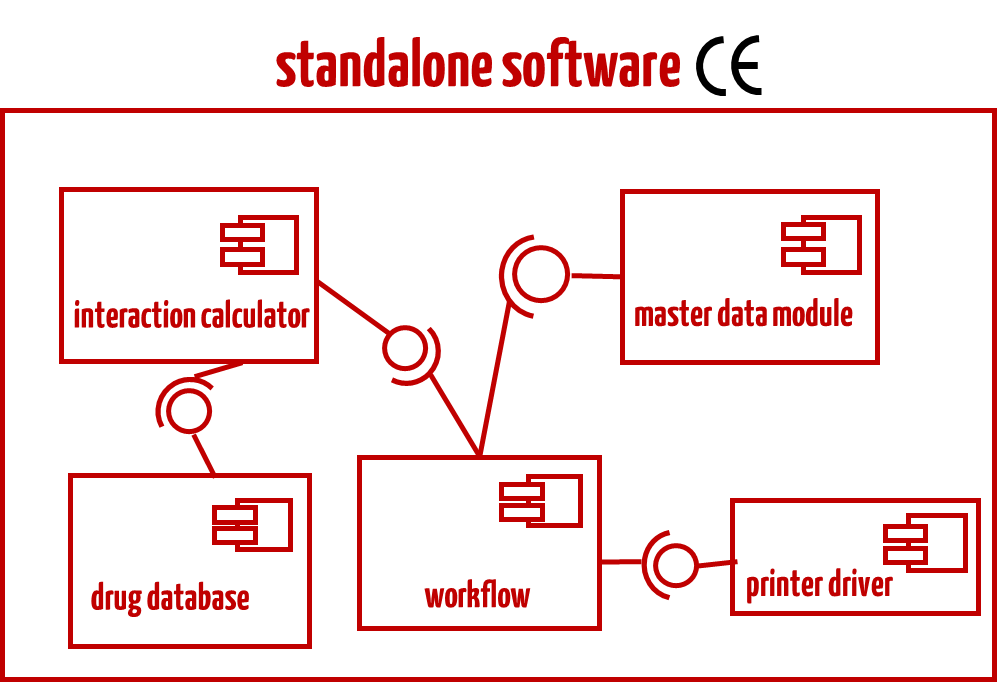
In most cases, the standalone software will be certified as a medical device, not its components. For example, you would not aim to certify individual modules for drug safety software.
2. Confirmation by the ECJ
An ECJ ruling (German) from December 2017 confirms the possibility of differentiating between software modules with/without medical device status. The European Court of Justice clearly states in paragraph 36:
“In the case of medical software which simultaneously comprises modules which meet the definition of ‘medical device’ and others which do not meet it and are not accessories within the meaning of Article 1(2)(b) of Directive 93/42, only the former modules fall within the scope of that directive and must be CE marked.”
The judges even interpret the MEDDEV 2.1/6 guideline as follows:
“These guidelines further state that software which does not interfere with the data in any way or which is limited to storage, archiving, lossless compression or ultimately simple searching, i.e., in this latter case software which has a database function and by means of which information derived from metadata can be found without modifying it or interpreting it, may not be considered a medical device.”
The judgment also states:
“In that regard, the Commission’s guidelines in Title 4 (‘Modules’) referred to in Paragraph 33 of the present judgment essentially confirm that where software is composed of modules which meet the definition of ‘medical device’ and those which do not, only the former need be CE marked since the others are not subject to the determinations of that Directive. These guidelines make it clear that it is up to the manufacturer to specify the limits and interfaces of the various modules. As far as the modules subject to Directive 93/42 are concerned, these must be clearly indicated by the manufacturer and be based on the future use of the device.
Therefore, the manufacturer of such software is obliged to specify the modules that constitute medical devices so that the CE marking can be affixed to these modules alone.”
You can download the judgment here (German).
Summary
- A software can consist of different modules, where only specific modules are to be certified as medical devices.
- The manufacturer must specify these modules and affix a CE marking to these modules.
- The manufacturer must clearly identify the boundaries between these modules, whereby the division must be based on the intended purpose (see page 18, penultimate paragraph of MDDEV 2.1/6).
The ruling should not be understood to mean that standalone software must always be broken down into modules, and then a decision must be made for each module as to whether it is a medical device. It is true that IEC 62304 requires software to be broken down into components (except for class A software). However, manufacturers should decide very carefully whether they actually have individual modules certified as medical devices. In most cases, the Johner Institute advises against this (see below and example in Fig. 3).
3. Evaluation
a) Legal certainty exists
Like some notified bodies, the Johner Institute thought that manufacturers could place a device on the market either as a medical device or as a non-medical device. It suspected that the authors of MEDDEV 2.1/6 understood terms such as “module” differently from software developers. Sentences such as the following reinforced the impression of a separate conceptual world:
“Computer programs(sic!) used in healthcare mostly have applications consisting of medical device and non-medical device modules.”
However, the ECJ judges followed the MEDDEV so that there is now clear case law. However, the judges did not define what a module is. Are they components, as understood by a software developer, that are an (indispensable) part of the software? Or are they plugins that can be installed additionally and optionally?
But you also have to understand the background of the ruling: The French state wants to regulate certain software. Philips does not want to allow it to do so because the device is CE-marked and, therefore, excluded from further regulation. The court says: In this case, the French state is right because the software is largely not a medical device. So specifically a device, a HIS, where really large parts, which are also separable, are not a medical device.
b) Advantages of the judgment
With the publication of the ruling, manufacturers of standalone software can use the option to certify only those software modules as medical devices with a corresponding intended purpose. This is recommended in the following situations:
- There are only a few or/and only small modules with a medical intended purpose compared to the entire software.
- The modules are organized according to (medical) functions and are sufficiently separated from each other.
- The effort required to certify the entire software is not affordable for the manufacturer.
The Johner Institute recognizes a number of reasons that speak in favor of the EC J’s decision:
- The decision may be helpful in terms of deregulation and as a counterbalance to the fatal Rule 11 of the MDR for manufacturers.
- It follows the trend that medical devices are increasingly opening up to “dissolving,” and operators are combining components (from different manufacturers) into systems.
- This forces manufacturers to think in terms of functional and weakly coupled components. This improves the software architecture and, thus, stability, maintainability, and testability.
c) Challenges and risks
However, manufacturers who take advantage of this option should be aware of the consequences:
- Risk management
In risk management, it may at least be difficult to argue that the risks are controlled if the data that a certified module benefits from originates from modules whose quality cannot be adequately assessed because the documentation of the respective software lifecycle (in accordance with IEC 62304) is not available. - Usability
A module’s usability cannot usually be assessed individually. It is questionable whether inspecting a “window,” a “screen,” or a mask (without considering the context of the entire software) provides reliable output. IEC 62366-1 requires usability to be assessed for use scenarios rather than individual technical modules or UI elements. - “Audit safety”
Software developers are becoming more susceptible to questions from auditors as to whether the code they have just developed belongs to a certified or non-certified module. Many development departments feel a sense of pride if they succeed in anchoring a single set of best practices… - Regulatory efforts
If several modules a manufacturer places on the market together are medical devices: does this even constitute a system or procedure pack? Is a corresponding declaration required? Then, the manufacturer would not have to register a device but modules and also a system or procedure pack. - Software lifecycle and documentation
The division into modules, each of which is an individual medical device, makes it possible to develop the modules independently. However, this requires separate documentation for each medical device, its intended purpose, risk management file, software life cycle file, declaration of conformity, manuals/instructions for use, clinical evaluation, etc. The challenges of usability engineering have already been discussed. - Labeling
The judges did not discuss where the CE marking can and should be affixed in the case of individual modules in accordance with the requirements of the MDR and MDR. How should this be done for a module without a user interface?
Note: If manufacturers do not want to create these documents for each module and thus multiple times, if they do not want to carry out and document post-market surveillance numerous times, they must continue to market the software as a single device. But what have they saved? They have created the complete technical documentation that is required for the approval of a medical device.
IEC 62304 allows limiting the software documentation to the “critical” modules/components. It is doubtful that a possibly simpler argument regarding the segregation of software items justifies the additional effort involved in placing individual modules on the market as medical devices.
The question remains whether the device surrounding the components will also receive a CE mark. The MDCG writes:
This raises the issue of whether the whole product can be CE marked when not all applications have a medical purpose.
MDCG 2019-11
d) A shift of the levels
Anyone who finds it absurd that a software module can be a medical device should remember the first discussions about whether standalone software can be a medical device. This is because standalone software cannot function without a platform (hardware, operating system, possibly a runtime environment) and requires the technical interfaces of the platform (monitor, keyboard, touch, data interfaces, etc.). No one now questions whether standalone software can be a medical device.
For a software module as a medical device, the boundary between the medical device and environment (platform) is shifted by another level.
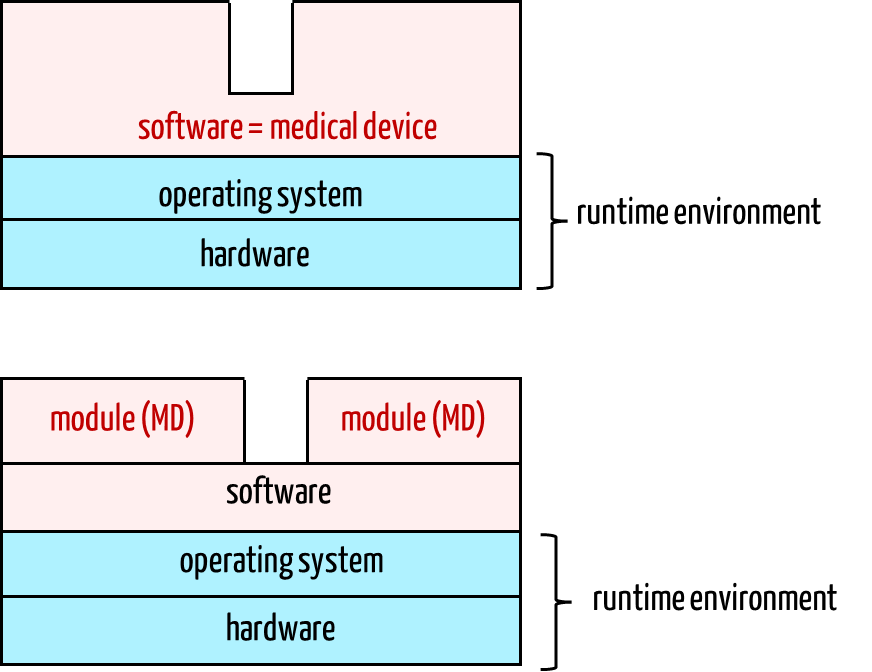
The module as a medical device now requires the proprietary software system as the first environment level, which in turn requires the operating system, and this, in turn, requires the hardware. The technical documentation for the modules (= medical devices) must specify this extended runtime environment.
During verification and testing, the software system must also be considered in the same way as for standalone software, just as the operating system and platform must be considered for the standalone software product.
e) Conclusion
The FDA started deregulation a long time ago. The fact that a comparable signal is (finally) coming from Europe is a good thing. The ECJ’s decision opens up new possibilities for manufacturers and makes combining devices and systems from modules easier.
Whether the effort required to approve one or more modules as a medical device is less than the effort needed to approve the entire software (which contains these modules) is questionable and must be examined on a case-by-case basis. As a rule, the path opened up by the ECJ is not advisable.
It is to be feared that the motivation behind the ruling has led to a decision that will cause more problems in practice than it will provide relief.
Change history:
- 2025-07-09: Links to new version of guideline MDCG 2019-11 added, section C) 5. (MEDDEV) and C) 12. (MDCG) revised
- 2025-02-19: Paragraph in C.2 and several links to official websites added
- 2023-11-17: Paragraph with ChatGPT updated due to notification from E-Health-Com
- 2023-02-03: Paragraph with ChatGPT added, editorial changes
- 2022-16: Digital Health Policy Navigator linked under “Decision aids”
- 2022-09: Current MHRA document added under C)
- 2022-09: Under C) 13 Guidance of the Australian TGA added
- 2022-08: Section “B) Qualification/Classification of software as a medical device” updated
- 2020-12: The outputs of the feedback process have been incorporated into chapter “13. Australia”
