Regulation (EU) 2025/327 on the European Health Data Space (EHDS for short) is another European regulation that may affect medical device and IVD manufacturers.
This article explains what this regulation requires and what these manufacturers have to do by when, as well as the possible advantages of the EHDS and the EHDS Regulation.
This article uses the terms “Regulation (EU) 2025/327” and “EHDS Regulation” synonymously.
1. Objectives and beneficiaries of the European Health Data Space (EHDS)
a. Overview
Various parties shall benefit from the European Health Data Space:
- Patients: Patients shall have better access to and control over their health data and benefit from better healthcare. A cross-border exchange of data also contributes to this.
- Healthcare providers: Hospitals, medical professionals such as physicians, and other medical service providers can access more complete data.
- Research: Research should also benefit from these data, enabling more innovation and better healthcare.
- Politics: Political decision-makers have access to reliable data, which allows for quick and targeted decisions, such as in a pandemic.
- European Union: The EHDS is essential in creating a strong and resilient European Health Union.
On its website about EHDS, the EU provides links to further sources of information, including a fact sheet that lists the advantages of European Data Spaces, including the economic advantages.
b. Advantages for medical device and IVD manufacturers
It is still unclear whether the manufacturers consider the uniform legal and technological framework and the interoperability required to be beneficial.
However, it is to be expected that the manufacturers will benefit from the new legal possibilities for using health data (“secondary use”) for
- “ensuring high levels of quality and safety” (Article 53 a)),
- “scientific research […] that ensures high levels of quality and safety […] of medical devices” (Article 53e), and
- “training, testing and evaluation of algorithms, including in medical devices, in vitro diagnostic medical devices, AI systems and digital health applications;” (Article 53e)
Thus, the EHDS will likely become an important source for post-market surveillance and post-market clinical follow-up.
Authorized access to further health data also enables manufacturers to develop new business models and evaluation options.
Medical devices can provide medical professionals with better support for diagnosis or therapy suggestions if these are based on a more comprehensive data set. The EHDS Regulation is an important pillar for personalized medicine.
Medical devices such as medical apps can use a wealth of data to support patients in preventing, alleviating, and curing diseases.
2. Who is affected by the EHDS Regulation
a. Overview
Regulation (EU) 2025/327 imposes obligations on various organizations. These include:
- Manufacturers of EHR (Electronic Health Record) systems
- Importers, authorized representatives, and distributors of these systems
- Medical device and IVD manufacturers, if their devices exchange data with EHR systems
- Manufacturers of wellness applications, if their devices exchange data with EHR systems
- Healthcare providers and healthcare professionals
- European and national (health) authorities
These groups are not mutually exclusive. For example, some EHR systems also count as medical devices.
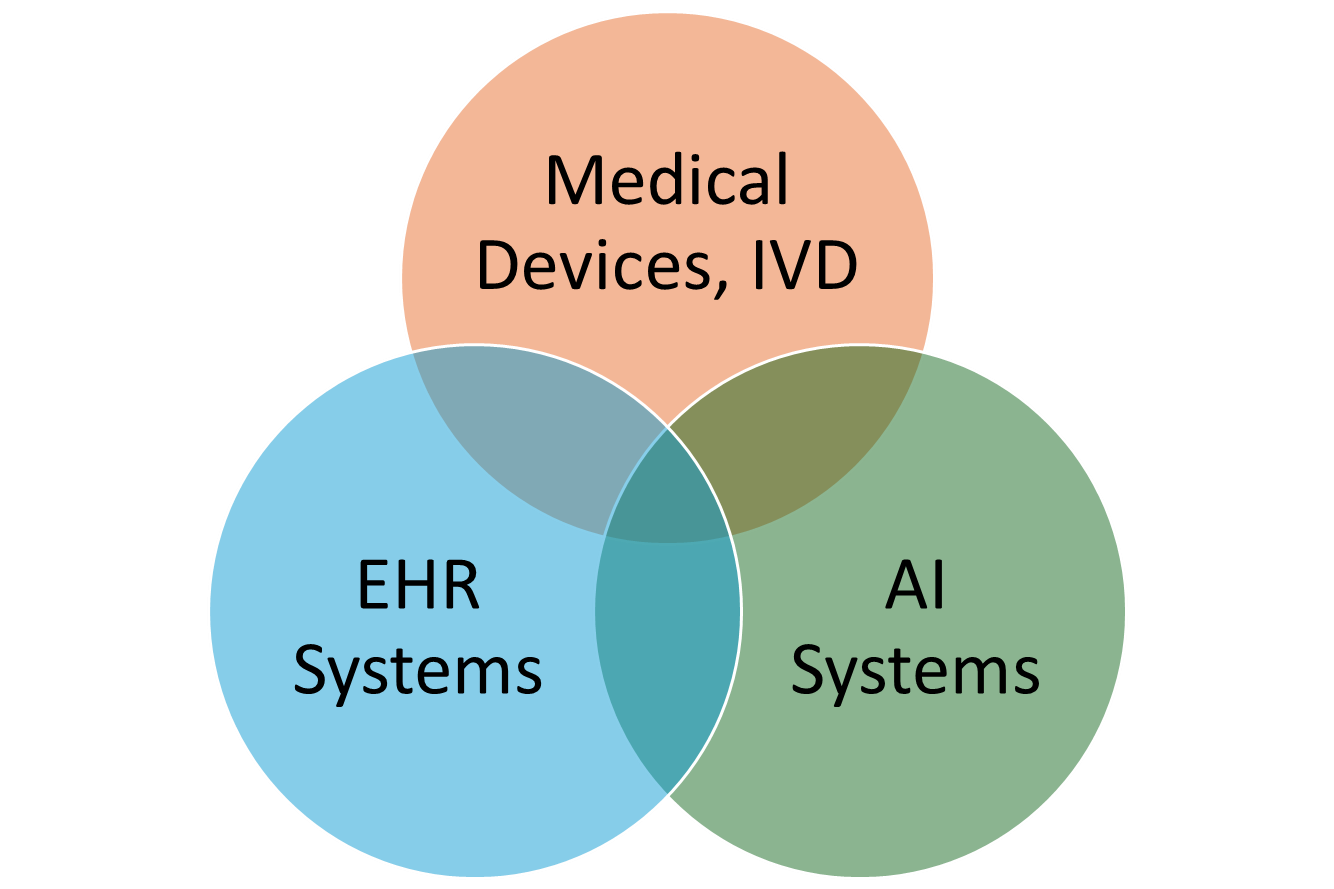
b. Manufacturers of EHR systems
The European Health Data Space Regulation defines the term “EHR system.”
any system whereby the software, or a combination of the hardware and the software of that system, allows personal electronic health data that belong to the priority categories of personal electronic health data established under this Regulation to be stored, intermediated, exported, imported, converted, edited or viewed, and intended by the manufacturer to be used by healthcare providers when providing patient care or by patients when accessing their electronic health data;
These “priority categories of personal electronic health data” are set out in Annex I. These include:
- “Patient summaries,” including demographic data, allergies, medical history, diagnoses, medical and care procedures, and medication
- Electronic prescriptions and dispensing of medicinal products
- Medical imaging and related findings
- Results of electronic examinations such as laboratory tests, including the associated findings
- Discharge reports
Typical examples of EHR systems are clinical information systems such as hospital, radiology, and laboratory information systems. EHR systems are also health apps used by patients if they allow access to electronic health data in accordance with their intended purpose.
c. Medical device and IVD manufacturers
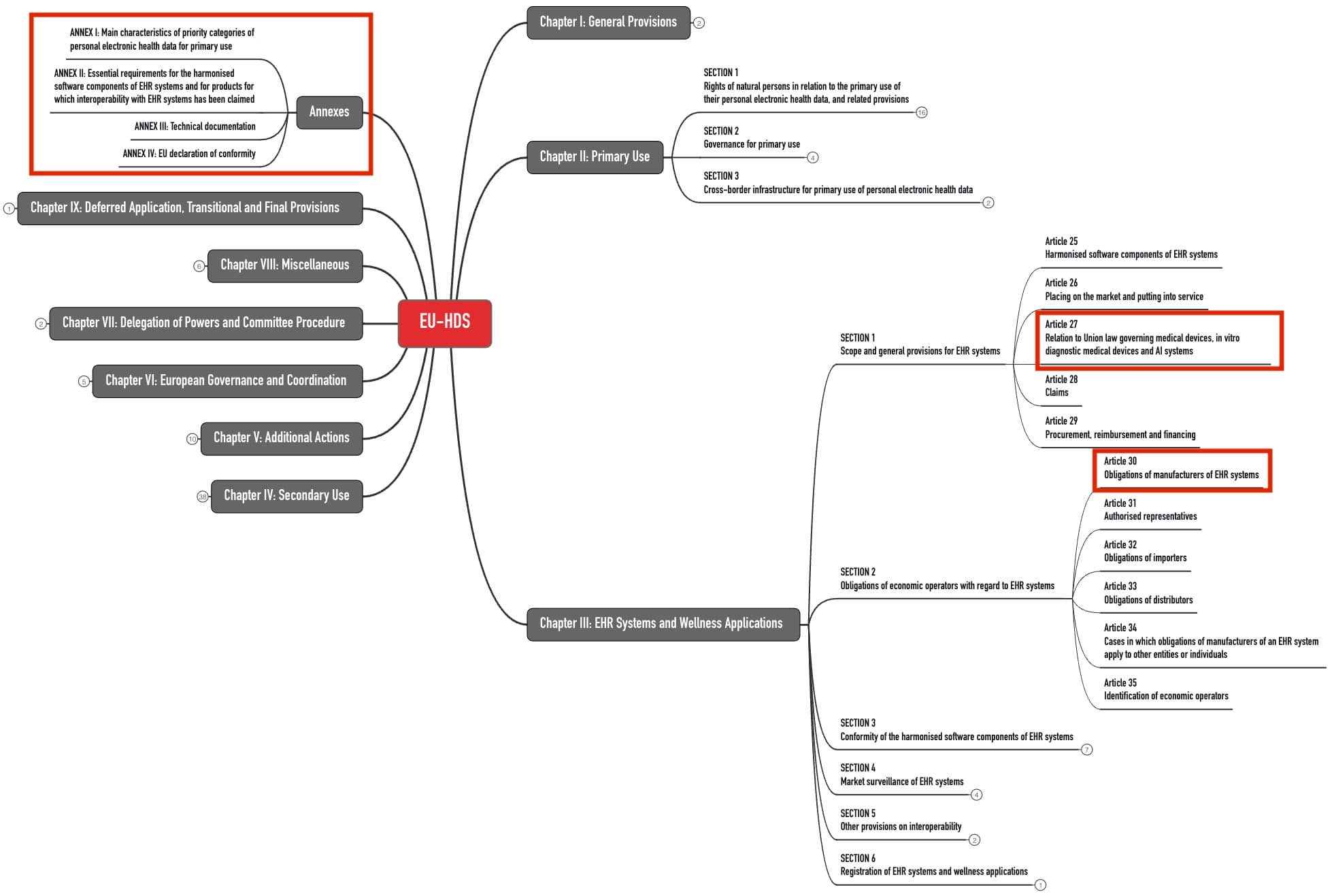
Medical device and IVD manufacturers are affected when they claim their devices are interoperable with EHR systems. Article 27 of the EHDS Regulation regulates this.
3. Obligations of EHR manufacturers
a. Interoperability of devices
The above-mentioned “priority categories of personal electronic health data” must be transferable via a machine-readable format between “different software applications, devices and healthcare providers” (Article 15). Inspection procedures are planned for this.
The Commission will set up a central interoperability platform (MyHealth@EU) for cross-border data exchange.
EHR manufacturers will have to integrate a “European interoperability software component” into their software.
a software component of the EHR system which provides and receives personal electronic health data under a priority category for primary use established under this Regulation in the European electronic health record exchange format provided for in this Regulation and which is independent of the European logging software component for EHR systems;
EHDS Regulation, Article 2(2) n)
The components of this interoperability software must meet the requirements of Annex II, in particular Section 2 thereof.
That includes requiring the software component to be able to send and receive health data in the standardized exchange format and not prohibit, restrict, or impede any permissible access.
b. Data protection and data security of the devices
In addition, the devices must meet data protection and IT security requirements. The EHDS Regulation supplements the “Cybersecurity Regulation” (EU) 2024/2847.
It requires EHR manufacturers to verify the essential requirements of the Cybersecurity Regulation (Annex I) as part of the conformity assessment according to the EHDS Regulation.
The data protection and security requirements mean that EHR systems must provide “reliable identification and authentication mechanisms,” which is also required by the EHDS Regulation in Annex II, Section 3.1.
In addition, the regulation requires a “European logging software component.”
a software component of the EHR system which provides logging information related to access by health professionals or other individuals to priority categories of personal electronic health data established under this Regulation, in the format defined in point 3.2. of Annex II thereto, and which is independent of the European interoperability software component for EHR systems;
EHDS Regulation, Article 2(2) o)
This component logs who accesses which data, when, and from where the data originated.
c. Further requirements
Further requirements for EHR system manufacturers will be familiar to medical device manufacturers:
| Requirements | Comments |
| Proof of compliance with the above-mentioned essential requirements | To this end, the EU plans to publish Common Specifications by March 26, 2027. |
| Technical documentation according to Article 37 and Annex III | By comparison, the requirements for the TD of medical devices are much more extensive. |
| Declaration of conformity | |
| CE marking | |
| Registration | If these devices are also medical devices or IVD, EUDAMED will be the database for registration. |
| Obligation to take corrective actions | This requires cooperation with the authorized representatives, distributors, and authorities. |
| Provision of the source code in justified cases | This is to enable authorities to check compliance with the essential requirements. |
The member states may define national requirements for conformity assessment (Article 41). However, the EHDS Regulation does not impose any special requirements for the conformity assessment procedure. In particular, there are no notified bodies to be involved in the conformity assessment of EHR systems.
The conformity assessment procedures under other EU regulations (in particular, MDR, IVDR, AI Act) remain unaffected.
4. Obligations of other actors
a. Obligations of other “EHR economic operators”
The obligations of other economic operators are comparable to those under the MDR and IVDR:
- Authorized representatives
- Importers
- Distributors
b. Obligations of medical device and IVD manufacturers
The medical device and IVD manufacturers affected by the EHDS Regulation under Article 27 must also comply with the essential requirements for the two software components:
- European interoperability software component for EHR systems
- European logging software component for EHR systems
Annex II Section 2 of the EHDS Regulation specifies these essential requirements described above.
Like the manufacturers of EHR systems, in the future, the medical device and IVD manufacturers must also comply with Common Specifications (CS), which the Commission intends to adopt by March 26, 2027. The scope of these CS is determined by Article 36.
An additional conformity assessment procedure is not planned for these devices. These manufacturers also do not have to register their devices in any other database.
c. Obligations of manufacturers of wellness applications
Wellness application manufacturers also fall within the scope of the EHDS Regulation if they claim that their devices are “interoperable with an EHR system.”
any software, or any combination of hardware and software, intended by the manufacturer to be used by a natural person, for the processing of electronic health data, specifically for providing information on the health of natural persons, or the delivery of care for purposes other than the provision of healthcare.
The requirements for these manufacturers are less stringent than those for medical device and IVD manufacturers. They concern:
- Labeling of this application (Article 47)
- Interoperability (Article 48)
- Registration (Article 49)
The EHDS Regulation does not require (additional) CE marking.
5. Tips for implementation
a. Clarify affectedness
As a manufacturer, you should first clarify whether the devices you have on the market or plan to launch fall under the scope of the EHDS Regulation.
To do this, you need to get an overview of all development projects and “qualify” all devices on the market and all planned devices. This may require you to specify the intended purpose of these devices more precisely.
b. Assess consequences
The requirements for manufacturers (medical devices, IVD, EHR systems) may seem manageable compared to other regulations. However, these requirements strongly affect the technical implementation of the systems. In the worst case, a re-development of devices will be necessary.
As a manufacturer, you should, therefore, assess which of your devices you can still prepare for compliance, particularly regarding interoperability and logging requirements, and what efforts this will take.
c. Consider transition periods
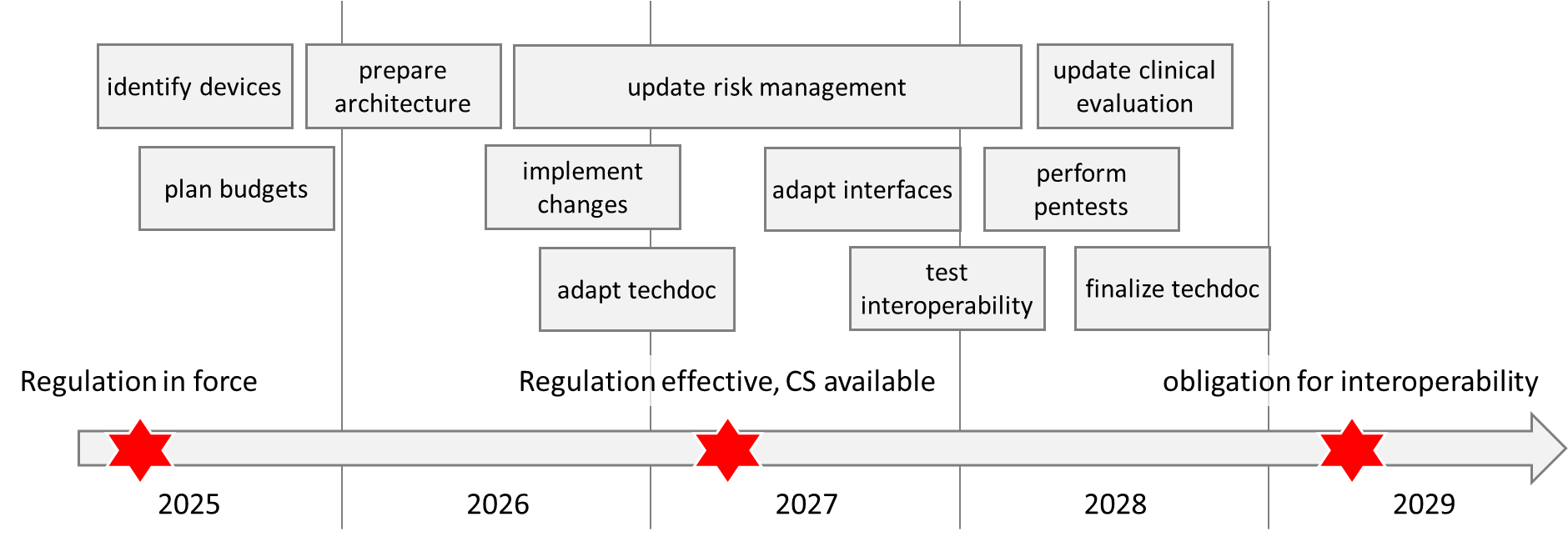
Before planning, you should take note of the transition periods:
The regulation came into force on March 26, 2025. It will apply from March 26, 2027.
However, it grants partial deferral to manufacturers of EHR systems and medical devices and IVD:
They will not have to comply with the requirements for software components and thus for interoperability regarding “priority categories of personal electronic health data” until 2029, and in the case of medical imaging or medical examination data (e.g., laboratory), not until 2031.
The regulation also grants a transitional period until 2031 for in-house developed EHR systems.
d. Plan projects
Once the costs and the latest completion date are known, you can start planning as a manufacturer. In doing so, you should consider the effects of the product changes:
- Revision of the data protection, role, authentication, and authorization concepts
- Changes to the software architecture and expansion to include the required components
- Analysis of how the modified interfaces affect interoperability with other devices
- Supplementing risk management
- Perform pen tests again
- Expanding post-market surveillance to include further data sources
Since many development projects take two or more years, it may be necessary to start implementation promptly, or in the worst case, it may already be overdue.
Don’t wait for the Common Specifications. There is no guarantee that they will be published by the promised deadline. Furthermore, there is a risk that the time between the deadline (March 2027) and the mandatory implementation (March 2029) will be too short.
e. Summary
Your timetable could look like the one in the following figure:
6. Conclusion and summary
a. Comprehensible objective
The desire to collect health data in a structured way and make it available in a European Health Data Space is understandable – whether for patient care (primary use) or research, statistics, and political decision-making (secondary use).
b. Possibly challenging implementation
Should more extensive changes to the devices and their software systems be necessary, the manufacturers are unlikely to find the transition periods very generous anymore. This is because the costs are high (see above).
Moreover, it is the details that are critical here:
- There is currently no Common Specification. If these do not appear until 2027, manufacturers will only have two years to comply.
- The obligation to log many actions, store data, and make it accessible can conflict with data protection requirements.
- Manufacturers must carefully consider how they formulate their devices’ intended use. The applicability of the EHDS Regulation depends on this.
- Devices can simultaneously count as an EHR system, a medical device, and a high-risk AI system. In this case, the requirements of three EU regulations overlap.
- The whole thing will only work if the EU and national authorities do their extensive homework. The history of EUDAMED and the Common Specifications for the MDR and IVDR raise skepticism.
c. Rapid clarification advisable
The above tips will help to quickly clarify the situation – including when implementation should begin at the latest. This way, manufacturers can better plan their development projects and allocate the necessary resources.
Clarify quickly whether and, if so, how you are affected. Then, plan the project and the necessary budgets. If you need help with this, please send me a short e-mail at info[at]johner-institute.com.
The Johner Institute also supports manufacturers in implementing Regulation 2025/327:
- Assessing which devices are affected by the regulation
- Evaluating software architecture and the impact of changes
- Defining an implementation plan
- Updating the risk management and clinical evaluation
- Assessing IT security, including pen testing


