1. Definitions
Medical software includes all software used for healthcare, particularly for medical devices or medical devices (embedded software), and software that is itself a medical device (standalone software).
IEC/CD1 82304-1 (Health Software – Part 1: General requirements for product safety) distinguishes between the following terms:
- HEALTH SOFTWARE
Software intended to be used specifically for maintaining or improving health of individual persons, or the delivery of care
- MEDICAL SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device or intended to be a SOFTWARE MEDICAL DEVICE
- SOFTWARE MEDICAL DEVICE
Software intended to be a medical device in its own right
- MEDICAL DEVICE SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device
This clarifies that medical software can be a medical device but does not have to be.
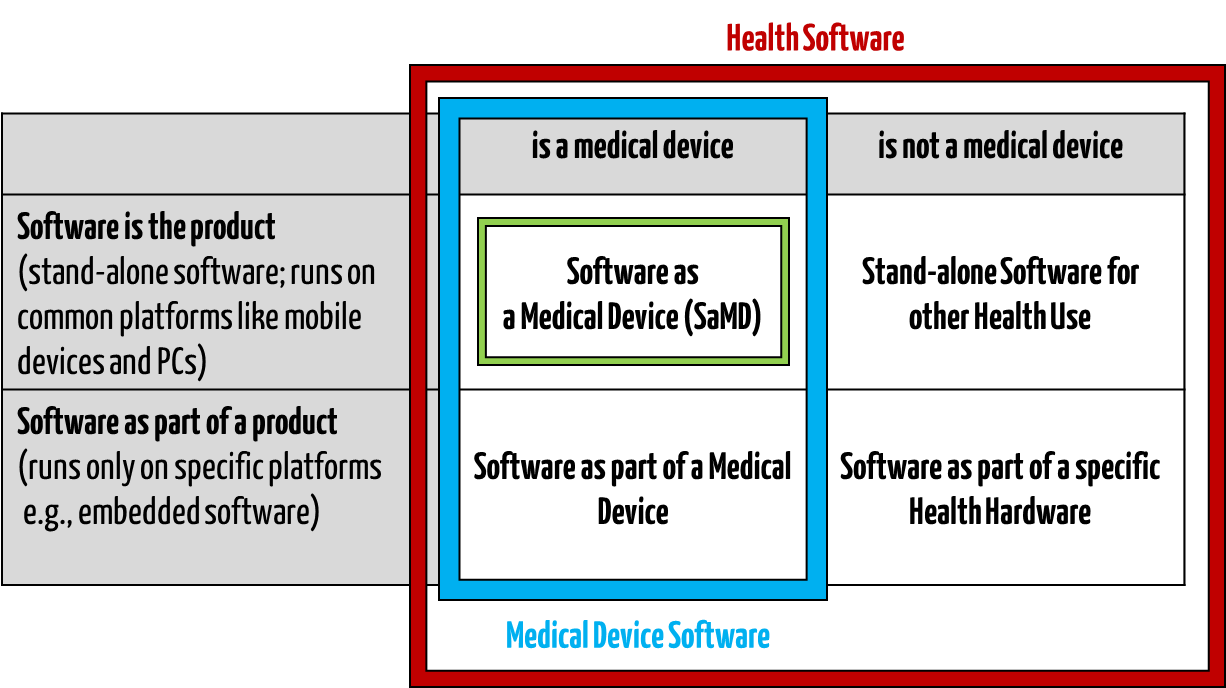
Fig. 1: Medical software includes medical device software and software as a medical device (click to enlarge).
2. Regulatory requirements
a) Medical software – a medical device?
The question often arises as to when medical software meets the definition of a medical device. You can find a further discussion on this topic in the article on the classification of software as a medical device and in the article on the qualification and classification of IVD medical device software.
b) Regulations, laws, standards
Software that is a medical device or part of a medical device must meet the regulatory requirements:
- In Europe, the medical device regulations (MDR, IVDR) are relevant. However, these only contain relatively general regulations for software, which this article presents.
- IEC 62304 defines the life cycle processes for medical device software.
- IEC 82304-1 applies to all “health software”. IEC 82304-1 also requires conformity with the requirements of IEC 62304.
- There are also MDCG guidelines, e.g., MDCG 2019-11 and MDCG 2023-4.
- The FDA sets out specific requirements in its guidance documents, including specific requirements for medical software. It also answered many questions specifically about software as a medical device in this FAQ.
3. Support for medical device manufacturers
Benefit from the support of the Johner Institute:
Contact us right away so that we can discuss the next steps. This will ensure that the “approval” is a success and that your software or devices are quickly launched on the market.
The cybersecurity standard IEC 81001-5-1 focuses on how IT security needs to be taken into account in the software life cycle. As a special standard for health software, it supplements IEC 82304-1 and IEC 62304 among others, and can close gaps that urgently need to be closed. The EU is currently planning to harmonize IEC…
Details
The Health Insurance Portability and Accountability Act (HIPAA) is a US law that establishes requirements for processing protected health data. Institutions that collect or process these data in the USA and their subcontractors must comply with HIPAA if they want to avoid sanctions. For European companies in particular, HIPAA is a regulation that is difficult to understand…
Details
We have known how vulnerable IT security is in the healthcare sector since February 2016, when the IT infrastructures of many clinics were brought to a standstill by a simple virus attack. As a result, the authorities are paying closer attention to ensuring that not only clinics but also manufacturers guarantee the IT security of…
Details
Threat modeling is a “mandatory” topic for all manufacturers of medical devices that contain or are software. This is because threat modeling is a structured process for the systematic analysis of IT security risks that auditors consider to be the “state of the art.”
More and more manufacturers are using machine learning libraries, such as scikit-learn, Tensorflow, and Keras, in their devices as a way to accelerate their research and development projects. However, not all manufacturers are fully aware of the regulatory requirements that they have to demonstrate compliance with when using machine learning libraries or how best to…
Details
In a guidance document, the FDA explains how to implement a software change in a regulatory-compliant manner. It describes when you need a new 510(k) submission (Premarket Notification) and when you “only” need to document the changes.
Details
What a lot of people understand by accessories is different from the definition of the term in the German Medical Device Act (MPG). This article gives you an overview of the definition of the term, the regulatory requirements, and typical questions.
Details
The parameterization of software – in this context, we can also talk about customizing or configuring software – often leads to discussion, e.g., regarding responsibilities and the differentiation to in-house production. This article gives manufacturers and their customers important advice on what to look out for when parameterizing software and how to avoid the usual pitfalls.
Details
On November 21, the Johner Institute, together with TÜV SÜD, TÜV Nord, and with the support of Dr. Heidenreich (Siemens), published a guideline on IT security specifically for medical device manufacturers.
IEC 82304 is now available. This is a good reason to take a closer look at this standard for “health software products.”