The system architecture describes the components of a (medical) device and how these components interact with each other via interfaces. For standalone software, the system architecture and software architecture coincide.
1. Documentation of the system architecture
a) Notation language
The documentation should reveal the individual components and their interaction. It has been proven that using standard notations such as UML or SysML is beneficial and that working in PowerPoint with undefined notation elements is not advisable.
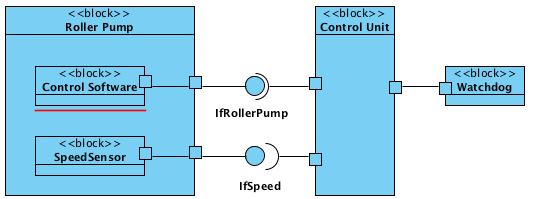
In addition to the diagrams with the different views, the interfaces of the individual components must be specified. Here, you will find information on how to describe interoperability requirements entirely.
b) Division of the documentation into system architecture and component specification
Do two documents need to be created: a system architecture and a component specification? Should these two documents be written promptly? By the same person?
There is no general answer to whether the system architecture and component specifications should be packed into one or more documents. It depends, for example, on
- how many components there are,
- how extensive the document would be,
- whether the components are to be developed by one or more independent teams,
- whether the component development is outsourced or not.
The component specifications should be made very promptly and in close coordination with the system architect. After all, they break down the system to be developed into components and describe how these components interact. In other words, they determine how the components behave via their interfaces in the sense of a black box. And that is exactly what a component specification is.

By the way, these considerations apply regardless of whether you are developing a medical device or standalone software; they are just as applicable to PEMS as to software systems. In one case, we are more within the scope of IEC 60601, and in the other, we are within the scope of IEC 62304.
2. Regulatory requirements for the system architecture
There are some regulatory requirements for the documentation of system architectures.
a) MDR and IVDR requirements
For example, the MDR (and analogously the IVDR) requires in Annex II:
a general description of the key functional elements, e.g. its parts/components (including software if appropriate), its formulation, its composition, its functionality and, where relevant, its qualitative and quantitative composition. Where appropriate, this shall include labelled pictorial representations (e.g. diagrams, photographs, and drawings), clearly indicating key parts/components, including sufficient explanation to understand the drawings and diagrams;
b) IEC 60601-1 requirements
IEC 60601-1 also requires that medical device manufacturers identify Programmable Electronic Subsystems (PESS), formulate requirements for them, and verify their fulfillment. Without a documented system architecture, these requirements cannot be met.
c) FDA requirements
The FDA’s requirements depend on the respective approval procedure. Even for 510(k) clearances, the FDA requires the following in this guidance document:
An overall description of the device design. A complete description of the device may be facilitated by the submission of engineering drawings or other figures. If the device consists of multiple components, a diagram identifying how the different components of the device system work together may be beneficial.
This corresponds exactly to the system architecture.
d) Further regulatory requirements
ISO 14971 does not explicitly require a documented system architecture. However, this is a necessary prerequisite for risk analysis and, in particular, for an FMEA.
A suitable system architecture, therefore, also serves the purpose of risk control.
In the Medical Device University, you will find video training on safety-critical system architectures.
3. Procedure for creating the system architecture
a) Decomposing the system architecture into components
When decomposing systems into components, some manufacturers tend to divide them into the “trades,” for example, mechanics, electromechanics, electronics, and software (left side in Fig. 3).
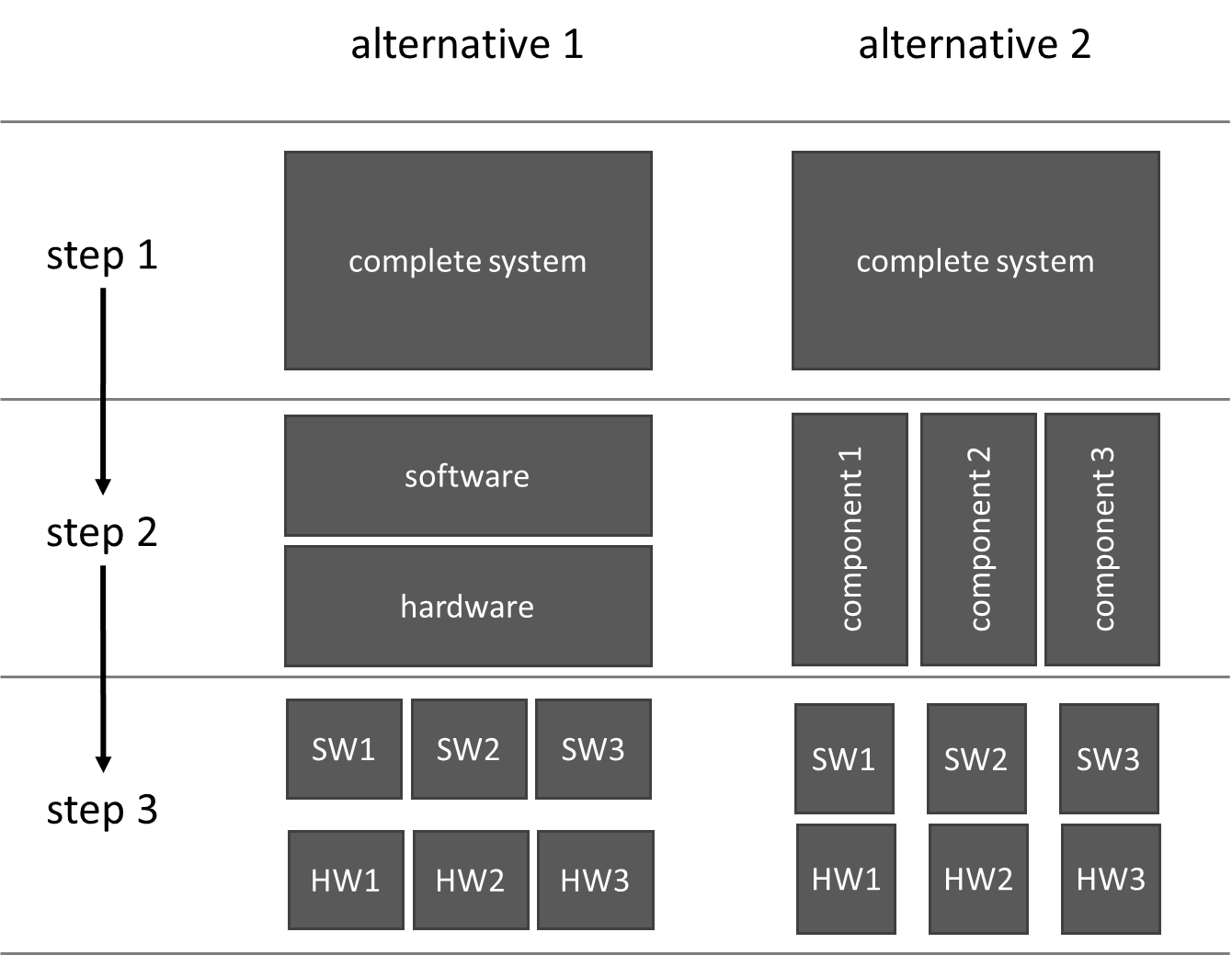
Many manufacturers have not had good experiences with this option. For one thing, this layered system architecture (hopefully) does not correspond to reality. For another, this “lasagna architecture” leads to a situation in which people do not consistently think in terms of (functional) components (right side in Fig. 3).
Only a consistently component-oriented system architecture leads to maintainable, safe, and testable medical devices and reusable components.
b) Interaction between system architecture and software architecture
For medical devices, it is not helpful to model software items at the first level of the system architecture. It is easy to see whether this is the case in the software architecture.
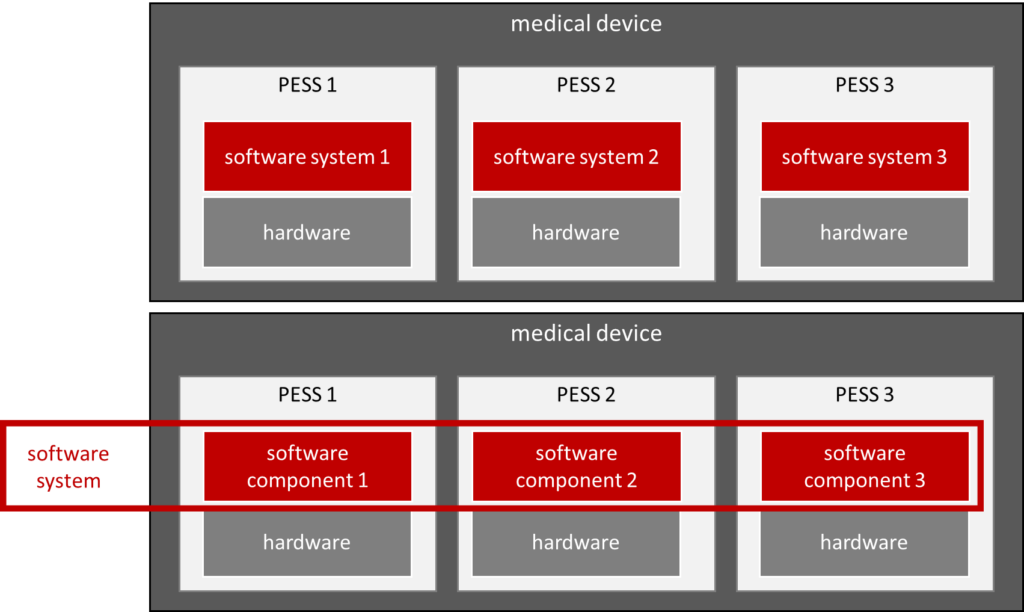
There are two variants for defining what the software system is.
Option 1: A software system is part of a PESS (upper part in Fig. 4)
If you define a software system as the totality of all software that runs in a memory/processor (and thus in a PESS), no software should be seen in the system architecture. (Unless the system contains a standalone software.) In the system architecture, you can then only see the components of which a subset is PESS. In each PESS, in turn, there is software (i.e., a software system) for which there are again software requirements and software architecture, and thus, software items are identified. However, the latter are not visible in the system architecture.
Option 2: A software system is the totality of all software (lower part in Fig. 4)
On the other hand, if a software system is understood to mean the entirety of all software in the medical device (the Johner Institute usually does not recommend this definition), then this would also be considered “lasagne architecture.” In other words, the manufacturer has not created a system architecture but rather a software architecture, an electronics architecture, and a “mechanical architecture.”
However, no legal or normative requirements exist for which variant you choose.
4. Summary and conclusion
Manufacturers of medical devices and IVDs usually need a system architecture. On the one hand, standards and laws require these system architectures. On the other hand, they are a prerequisite for the rapid development of safe, maintainable, and testable medical devices.
Therefore, manufacturers should take their time and ensure the necessary competences in the development team in order to develop the best possible system architecture. Confucius’ idea applies here: if you want to be fast at the end, you have to go slowly at the beginning.
The team at the Johner Institute specializes in creating and testing system architectures with and for medical device manufacturers. This allows to
- identify and control risks posed by the medical device,
- avoid difficulties in the audit and during approval,
- document system architectures in a way that is lean, precise, meaningful, and standards-compliant,
- find and correct errors early, thus avoiding expensive rework and project delays.
Change history:
- 2024-10-09: Article thoroughly revised, updated, and restructured. Images revised and added.
- 2015-04-23: First version published

