FMEA, or Failure Mode and Effect Analysis, is a procedure for investigating the unknown effects of known causes.
In the case of medical devices, for example, FMEA is used in risk analysis to analyze the consequences of a faulty component, in particular, the resulting hazards.
1. How to use the FMEA
a) FMEA in development
Use the Failure Mode and Effect Analysis to identify previously unknown hazards resulting from hypothetical failures:
- Start this analysis with the inputs and components of the device.
- Note possible failure modes for these inputs and components.
- Find out what effects (e.g., product misbehavior or hazards) these error states can have.
Let the example be a ventilator:
- Component: Software of the central control unit
- Error state of the component: Software crashes if sensors deliver unexpected values.
- Effects/hazards/risk: The crashed software brings the ventilator to a standstill, i.e., ventilation stops (hazard and hazardous situation). There is a certain probability that the patient will die (severity of harm). This is the risk.
Read more about hazard and hazardous situation and severity of harm.
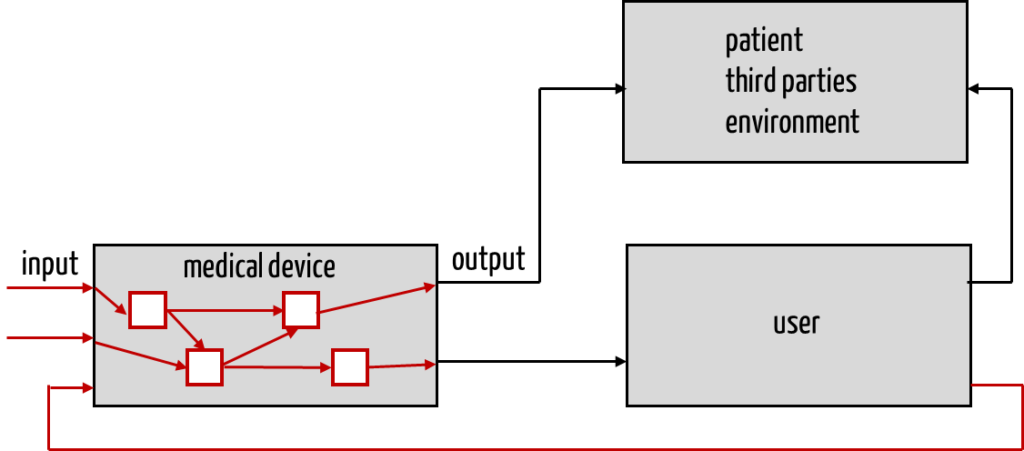
The method is called a bottom-up procedure because of the path from cause to effect.
b) FMEA when replacing or modifying components
As a medical device manufacturer, you should always use FMEA as the preferred procedure for risk analysis (more precisely, hazard analysis) to analyze the effects that replacing or modifying a component may entail.
You should also use the FMEA if you need to read the release notes and bug reports of your SOUPs and evaluate the possible consequences of these errors. By the way, this is required by IEC 62304.
It is also possible to apply FMEA to individual components that consist of subcomponents.
Component FMEA is not a risk analysis!
However, this form of FMEA is not used for risk or hazard analysis because the “component FMEA” does not discuss hazards as effects but rather the component’s misbehavior at its “outer edge.” As with the medical device itself, this misbehavior can be caused by a fault within the component or a faulty input to this component.
In other words, the component FMEA is not a risk analysis because it does not examine consequences in terms of hazards or harm but in terms of a faulty component. It can, therefore, only analyze the external failures and their probabilities.
c) FMEA in production
The same applies to the Failure Mode and Effect Analysis in production: This is also not a risk analysis procedure because it also does not examine harm or probabilities as a result of production errors, but instead errors in the produced device or a component of the device (as a result of a production error).
In the process or production FMEA, the manufacturer examines the process or production step by process or production step. The effects of possible errors are analyzed for each of these steps. Possible “externally visible” errors are
- incorrect physical, chemical, or biological nature of the component,
- component passes on information or data in an unspecified manner (too little, too much, too early, too late, wrong sequence, etc.),
- component releases energy or materials in an unspecified manner (too little, too much, too early, too late, wrong location, etc.).
Knowing the probability of which errors in the device will result in faulty production helps to estimate the probabilities of error in a component more accurately as part of the development FMEA.
Production consists of one or more processes. Read more about process FMEA (pFMEA) here.
d) FMEA as part of the QM system
In addition to production, the QM system determines other processes and procedures organizations use to develop devices and services and make them available to customers. Accordingly, the Failure Mode and Effect Analysis can be transferred to other processes.
It answers the question “What if…?”, i.e., if a process step or activity does not take place according to the specifications or does not deliver the desired output for another reason.
The method traces the chain of causes to the process outcome, possibly even further to risks for patients, users, and third parties or risks for the company’s compliance.
2. Tips for using the FMEA
a) Fulfill essential requirements: A documented system or software architecture
In contrast to PHA, FMEA, therefore, requires a system architecture or software architecture. You only know the individual components once you have documented these architectures. Only with an architecture can you understand how a fault in one of these components affects the overall system (medical device).
However, the architectures should not only show which other components a component interacts with but also how this interaction takes place. To be able to apply an FMEA, you, therefore need to know which
- data/information,
- materials or substances, or
- energies
one component can pass on to the next. In the case of “data interfaces,” you should be able to find this information in the component specification.
b) Document results in tabular form
Medical device manufacturers usually document the risks in tabular form and call this table the “FMEA table,” which is not entirely accurate. This term is misleading because the table is suitable for documenting the results of all risk analysis procedures (e.g., also those of the Fault Tree Analysis, FTA). This table usually has the following columns:
- ID
- Component/Input
- Fault in component/Input
- Internal chain of causes
- External misbehavior
- Hazard
- Probability of harm
- Severity of harm
- Risk before measures
- etc.
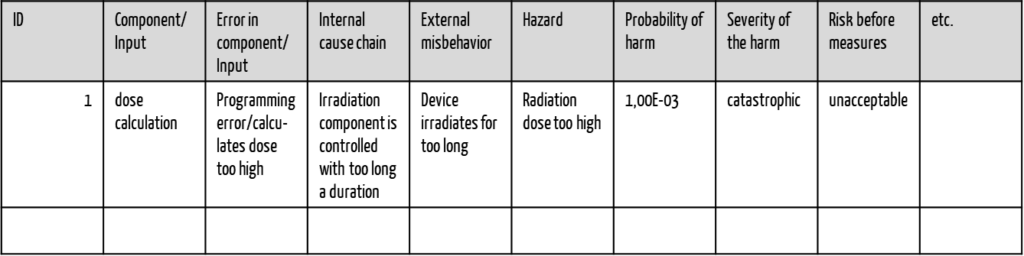
Manufacturers also usually document the risk control measures in this table.
c) Combine FMEA with HAZOP
FMEA can be combined with HAZOP analysis to identify possible input or component failures systematically. This procedure is described in IEC 61882 and works with guiding questions such as “too early?” “wrong sequence?” “too fast?” etc.
3. Advantages and disadvantages of the FMEA
a) Advantages
The FMEA has the advantage of being a very systematic procedure. Each component can be examined and “ticked off” for a given system or software architecture.
This procedure is easy to understand and corresponds to the mindset of many developers, who can start with “their” component.
b) Disadvantages
The FMEA is unsuitable for investigating and describing logical links between many faults. The Fault Tree Analysis (FTA) is suitable for this purpose.
It is also impossible to assess how granularly components need to be analyzed based on an FMEA. Many medical device manufacturers suffocate in “risk tables” with hundreds and thousands of lines.
Because developers are good at applying this method, they are often entrusted with the complete risk analysis. However, developers – in their role as developers – are neither qualified nor suitable for this.
Many manufacturers apply the FMEA using the Risk Priority Number RPN. This definition of risk does not match that of ISO 14971.
4. Conclusion and summary
The Failure Mode and Effect Analysis (FMEA) is a method that should be used in risk management. ISO 14971 presents this method as one of the most important.
FMEA is used and referred to differently in various industries. In the automotive industry, the identified “failure modes” serve as input for the actual risk analysis. In the medical device world, on the other hand, FMEA is often used as a method to identify risks and not just “failure modes.”
Regardless of the approach taken by manufacturers, they must trace the chain of causes to the actual harm and estimate their potential severity and probabilities. By definition, these are the risks.
The Medical Device University’s video training courses will teach you how to systematically create and document a risk analysis (including FMEA). The premium version contains ready-to-use templates.
Change history:
- 2025-05-14: Minor editorial changes such as missing links added
- 2023-10-02: Example in chapter 1, chapter 1.d), and summary (chapter 4) added; editorial changes
- 2020-10-20: First version created