Standards and laws require medical device manufacturers to take measures to control and minimize risks.
Content
This page briefly introduces this topic and links to further articles with practical tips.
- Measures for risk control
- Risk control in the product life cycle
- Special cases
- Support
1. Risk control measures
a) Objectives of the measures
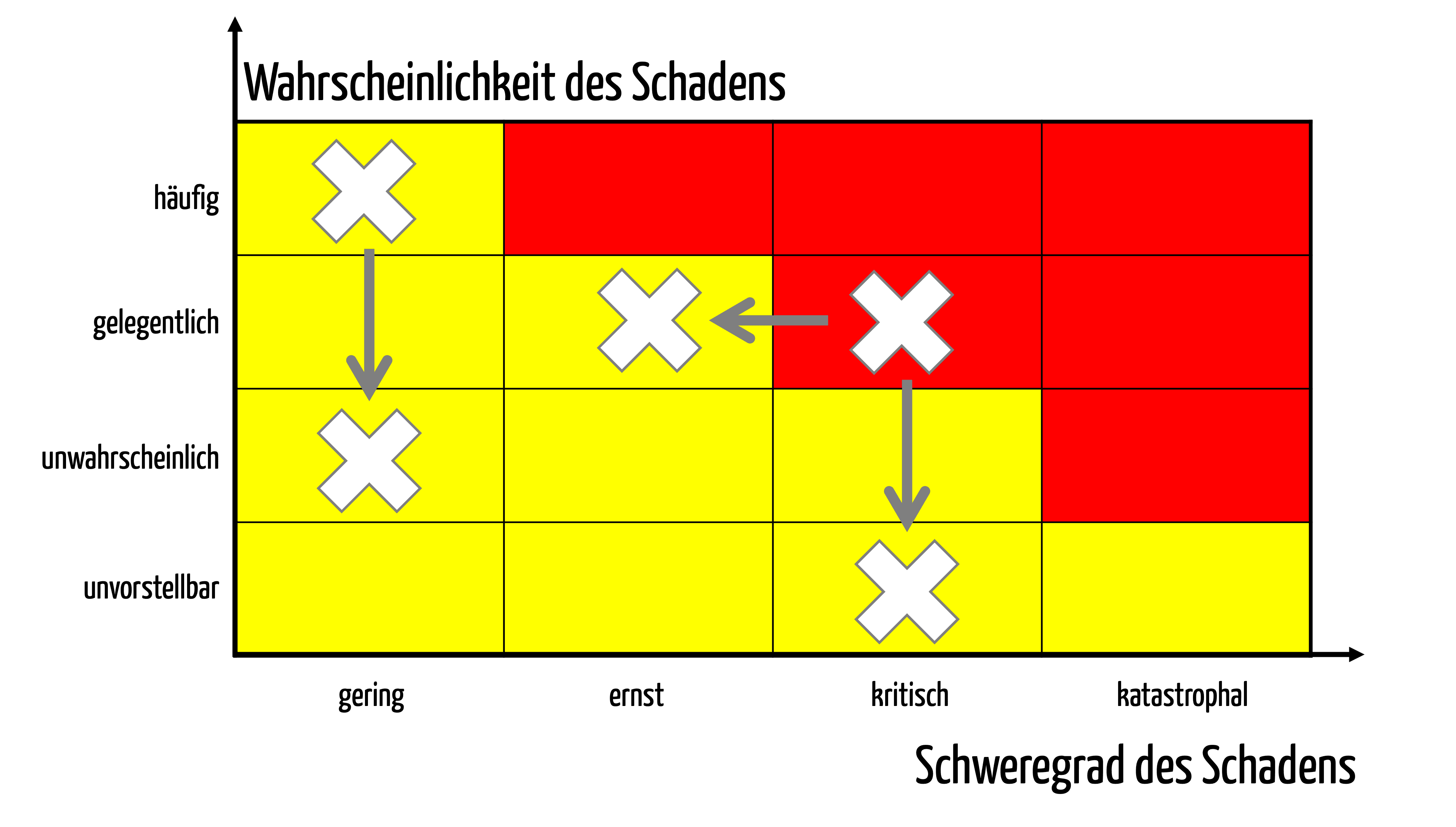
Manufacturers must not only control the risks but also minimize them to such an extent (see Fig. 1) that the residual risks are acceptable.
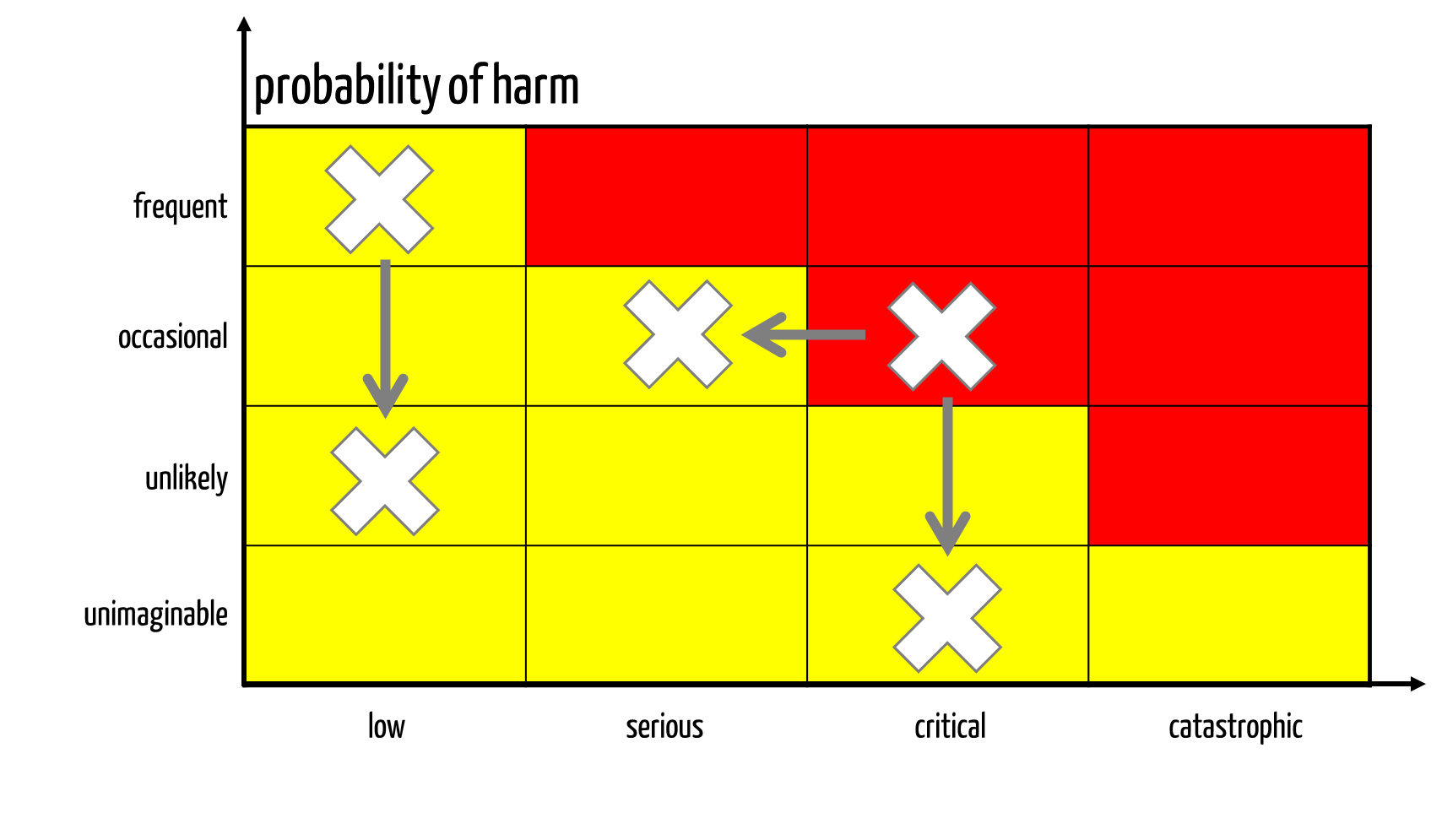
Fig. 1: Risk minimization measures reduce possible harm’s probability and/or severity.
The obligation to minimize risks applies to all risks, not just risks the manufacturer has assessed as unacceptable according to its acceptance criteria (red area in Fig. 1).
b) Types of measures
The laws and standards distinguish between three classes of risk minimization measures:
| Measure |
Explanation |
Examples |
| Inherent safety |
A device or process is designed to prevent risk from occurring. |
A device containing no allergic substances cannot trigger allergic reactions.
No one can get an “electric shock” from a device that is powered by a battery instead of mains power. |
| Means of protection |
Measures that reduce the probability or severity of possible harm |
A suspension that can carry eight times the required weight will rarely break.
A heterogeneous, two-channel architecture is less likely to cause harm if a component fails. |
| Information |
Guidance on the correct use of the device on the user interface, in the instructions for use, or during training courses |
A pop-up warning that two of the prescribed medications reinforce each other |
Tab. 1: The different types of measures
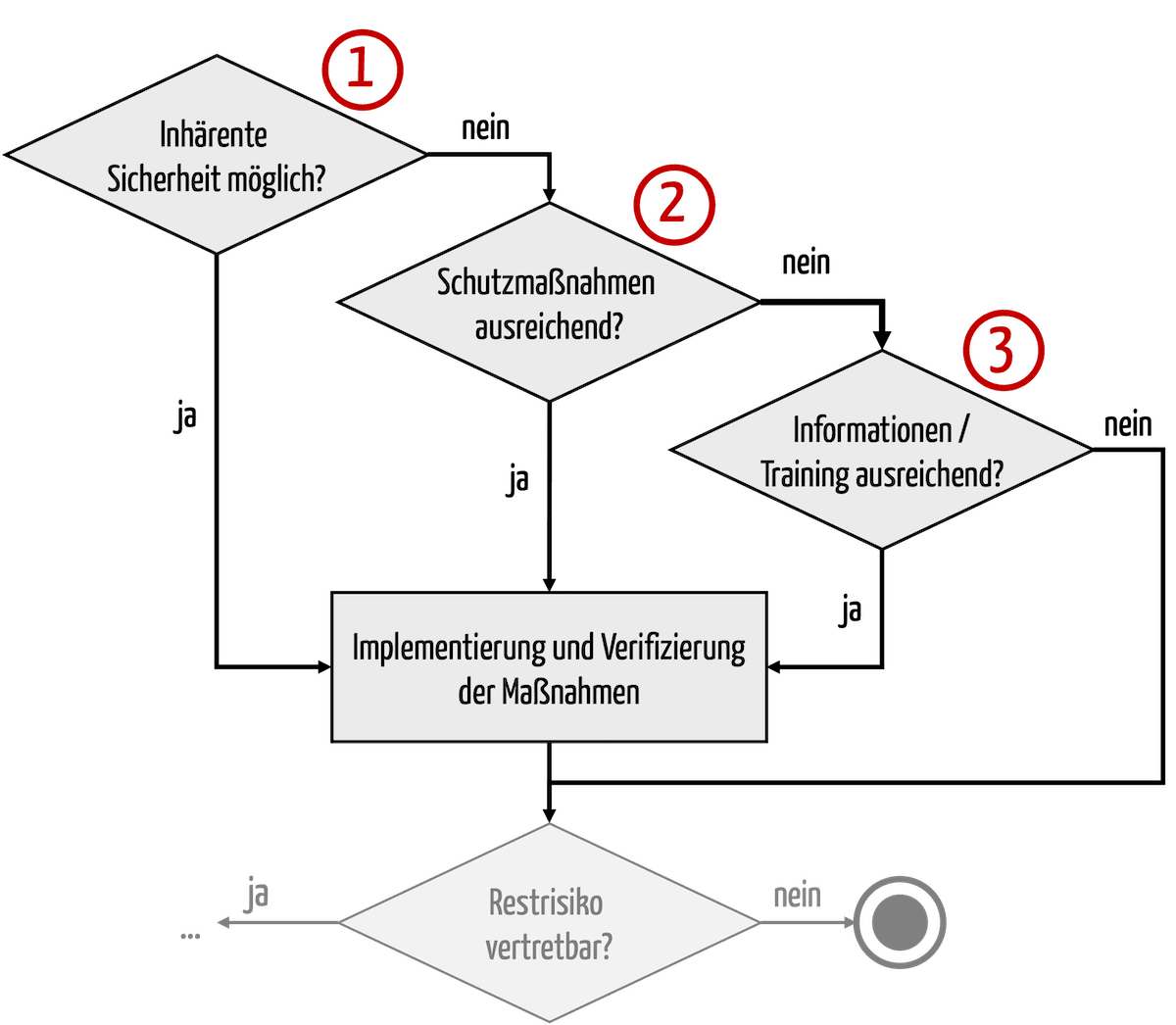
Standards and laws require manufacturers to implement the above measures in the order listed, i.e., to strive for inherent safety first (see Fig. 1).
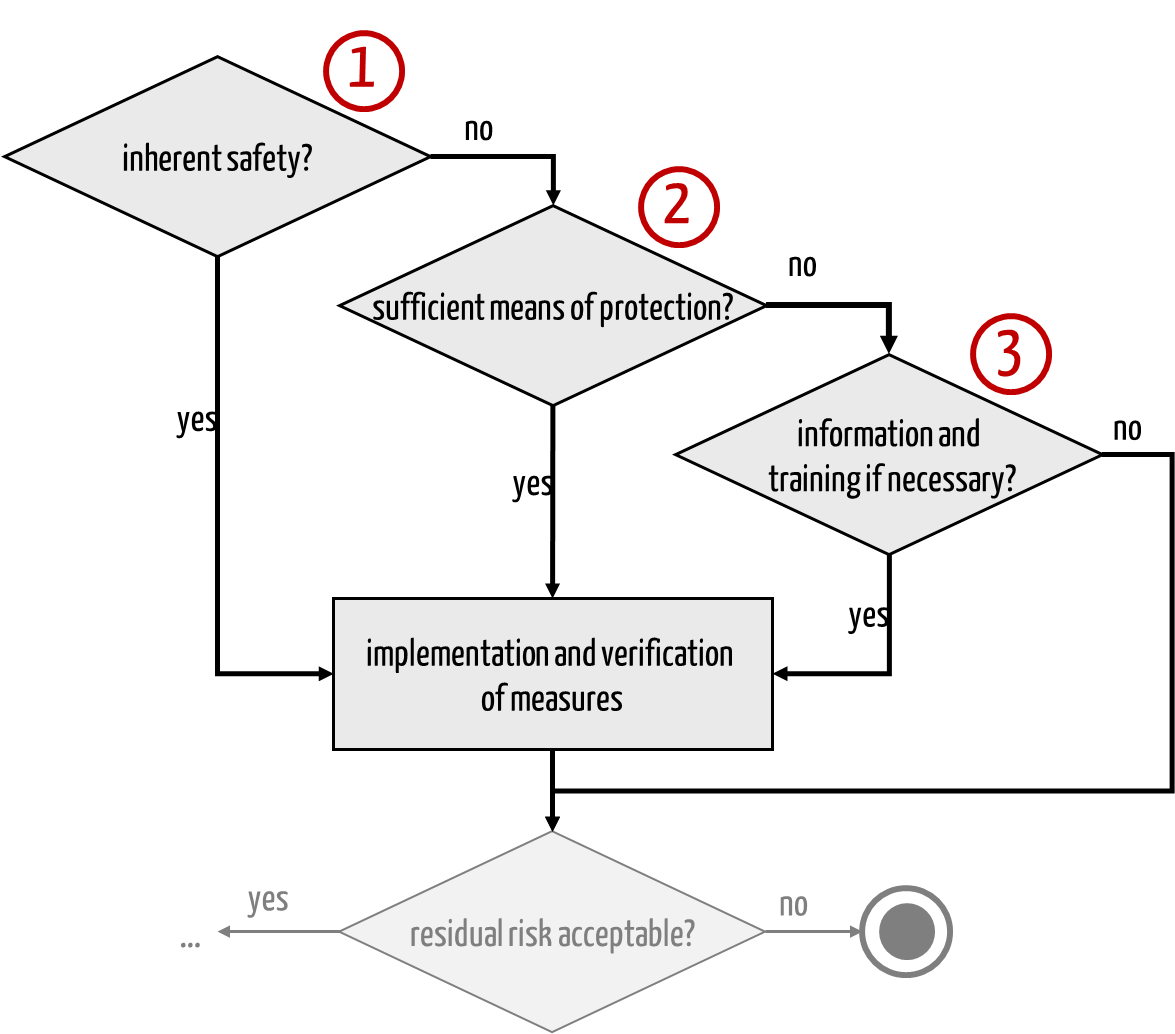
Fig. 2: The standards and laws fix the order in which risk-minimizing measures are implemented.
Caution!
It is wrong that information may no longer be used as a risk-minimizing measure. Read more about it in this article.
c) Regulatory requirements for the measures
Legislation in all markets and all relevant standards require risk control. Examples are
Manufacturers must document the implemented measures and proof of their effectiveness in the risk management report.
2. Risk control in the product life cycle
a) Determining the measures
The obligation to minimize and control risks is not limited to the development of devices but also applies to their production and operation, i.e., all phases of the product life cycle.
| Phases in the product life cycle |
Possible measures |
| Defining the intended purpose and identifying stakeholder requirements |
Restricting the intended purpose (e.g., the intended indications or patient and user groups) |
| Specification of the system requirements |
Specifications for the use of materials
Formative evaluation to minimize usability risks
Definition of a development process |
| Systems engineering and system architecture |
Use of components with characteristics of high safety (terminology of IEC 60601-1)
Functionally safe architecture (e.g., multi-channel architecture)
Use of established software libraries
Segregation of components
A system that goes into a safety state in the event of a malfunction |
| Production |
Specifications for the use of auxiliary materials
Restriction of production processes (e.g., components milled from solid materials are less likely to contain air bubbles than injection-molded parts)
ESD workstation for the assembly of circuit boards |
| Maintenance, servicing, operation |
Use only by the manufacturer’s trained service technicians
Regular cleaning of the device
Timely replacement of wearing parts |
Tab. 2: Phases in the product life cycle and possible risk control measures
b) Review of the measures
Manufacturers are obliged to check (e.g., verify and validate) the risk-minimizing measures in several respects:
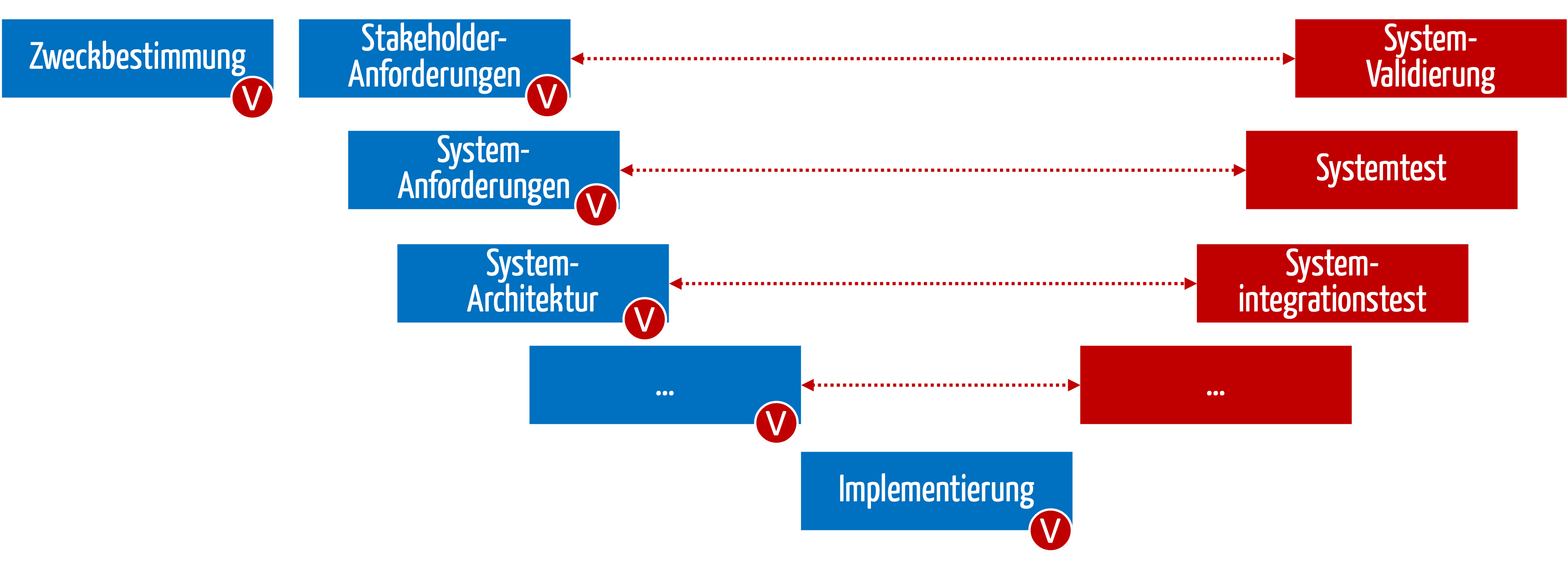
- Verification of documents
The (specification) documents created during development and production contain the measures (shown in blue in Fig. 3). The manufacturers must verify these documents (red V).
- Verification of the existence of the measures
Manufacturers must check whether the measures have actually been planned and implemented (red V).
- Validation/verification of the effectiveness of the measures
Finally, manufacturers must check the effectiveness of the measures (phases marked in red).
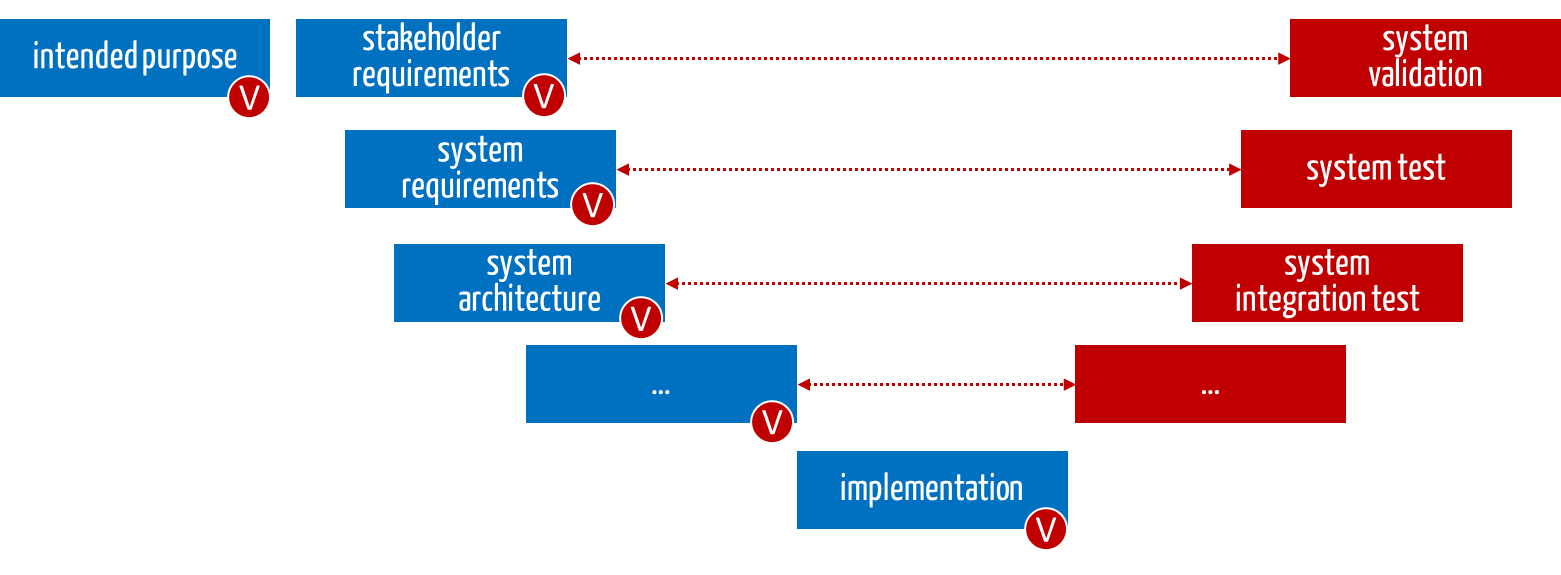
Fig. 3: The measures must be planned (in blue) and checked for existence and effectiveness (in red).
Tip
Safety assurance cases serve to prove the effectiveness of the measures and, thus, the safety of the devices.
4. Special cases
a) Testing
Many manufacturers list tests in the “risk table” as risk-minimizing measures. However, tests do not reduce the probability or severity of harm.
However, they help identify and eliminate errors (during development and production). These corrections and corrective actions reduce the risks.
Caution!
Tests do not lead to inherent safety, nor are they means of protection or information to minimize risk. Instead, tests make it possible to estimate the probability better and justify why it is as low as possible. This is because tests correspond to experiments or samples.
The task of tests is also to review the implementation and effectiveness of risk minimization measures.
b) Information
Information is permitted as a risk-minimizing measure, as this article explains.
The contrary assessment is due to a misleading formulation in the “Z Annex” of ISO 14971:2012, which is harmonized under the EU directives.
5. Support
Do you still have questions about risk-minimizing measures? You can get answers in our free micro-consulting.
In the risk management and 14971 seminar, you will learn about the regulatory requirements and how to implement them.
Benefit from the know-how of the experts at the Johner Institute to
Contact us right away to discuss the next steps. In this way, you can achieve safety devices quickly, without unnecessary effort, and promptly obtain approval.