In a guidance document, the FDA explains how to implement a software change in a regulatory-compliant manner.
It describes when you need a new 510(k) submission (Premarket Notification) and when you “only” need to document the changes.
1. What a software change is
a) Software changes in the “scope” of the guidance document
By a “software change” that falls within the scope of the guidance document, the FDA means any change to software that has already been cleared as standalone software or as part of a device (embedded software) under a 510(k) procedure.
This means that the guidance document does not apply to changes to software during development, nor does it apply to software that has undergone another approval procedure (e.g., PMA).
b) Examples of software changes
Examples of software changes are:
- Changed algorithm to increase benefits for patients further
- Improvement of the GUI (user interface), e.g., to minimize risks
- Replacement of a third-party component with the latest version
- Implementation of a new risk control measure
- Implementation of a new customer request, e.g., in the form of additional functionality
- Troubleshooting (bug fix)
- Refactoring to improve maintainability
- Switching to the next version of the programming language
c) IEC 62304 and software changes
IEC 62304 also prescribes what manufacturers should consider when making software changes. However, these requirements, which you can find out more about below, do not concern whether a new submission (e.g., to a notified body) is necessary.
2. When you need to resubmit to the FDA
a) The rules
The FDA always requires a new submission for changes unless one of the following conditions is met (the numbers refer to the chart below):
- (1) The change is solely for the improvement of IT security.
- (2) The change is purely a bug fix.
- There are no other objections.
You can consider the third point to be fulfilled if all(!) of the following conditions apply:
- (3) There are no new causes of errors, i.e., elements in a chain of causes that could lead to significant harm.
- (4) No new hazards or hazardous situations could lead to significant harm.
- (5) The change does not affect an existing risk control measure or require such a measure.
- (6) The change cannot impair the (clinical) performance and fulfillment of the intended use.
- (7) Other changes are controlled.
Once again, there is an ambiguous paragraph (point 7), which we will examine in a moment.
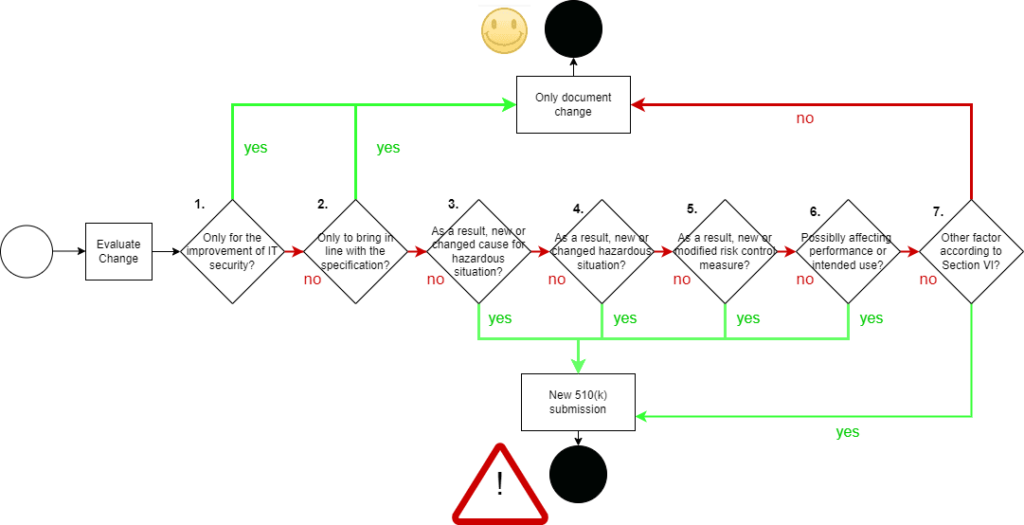
The illustration clarifies that the FDA makes the need for resubmission dependent on whether additional or higher risks may arise from the software change. At the same time, the FDA does not want to impede improvements in IT security through bureaucratic hurdles.
b) Examples
The numbering in the list above corresponds to that of the FDA. The FDA gives examples for each point, which we have taken up, supplemented, or reproduced here in more understandable language.
- Improvement of IT security
- Checking of user input to an application for cross-side scripting
- Replacing the encryption algorithm with a safer version
- Bug fixing (bringing the software in line with its specification)
- Replacement of the incorrect less-than sign (<) with the correct less-than-equal sign (≤)
- Addition of the forgotten masking of special characters when reading HL7 messages
- New error sources
- Addition of a new treatment parameter for an EEG device
- Provision of support for an additional operating system
- New hazards or hazardous situations
- An implanted active medical device that contains software should be able to be configured “from the outside” via a wireless connection. This new transmission can mean an energy supply and thus a new or changed hazard (situation).
- New or changed risk control measures
- A surgical robot’s alarm limits are expanded to minimize false alarms that occur in practice.
- When reviewing a sample number, the software no longer requires the operator to release the measurement explicitly but only issues a warning and logs the problem.
- Possible impact on performance or achievement of the intended use
- To increase the throughput of an IVD medical device, the manufacturer changes the software to shorten the measurement time.
- The manufacturer wants to improve the detection of arrhythmias from ECG data with a new software algorithm.
- Other factors: There are factors for which none of the above criteria apply but for which a new submission must still be considered. The FDA encourages manufacturers in this gray area (as it calls it) to get in touch. It cites the following as examples:
- Changes to programming language
- Porting to new hardware, operating systems, middleware, etc.
- Reengineering and refactoring
Change requests are usually formalized and documented requests or requests to change a device or software. In the case of medical devices, change requests must comply with regulatory requirements.
3. Change requests
a) What are change requests?
As the name suggests, change requests are requests from customers, users, or employees to change something
- to the device or the code,
- to the configuration/parameterization of the device,
- to the environment in which the device runs.
Most medical device manufacturers do not differentiate between user requests and user requirements when it comes to change requests from users. In most cases, user requests correspond to specific (new or changed) system/software requirements or system/software specifications and not user requirements.
Read more about the difference between user/customer request and user/customer requirement (German).
Manufacturers should distinguish between the two in their ticket systems, just as the FDA and ISO 13485 distinguish between stakeholder requirements (this includes user requirements) and system requirements.
Manufacturers are also obliged to make a distinction between:
- Corrections
- Corrective actions
- Preventive actions
- Other product improvements
Read more about corrections, corrective actions, and preventive actions.
b) Change requests and risk management
Many companies work with ticket or ALM systems, which they use to map the complete workflow, from the first “customer request” to the device’s release to the subsequent change request. It is sometimes noticeable that the tickets have a “risk classification” in the form of a drop-down list, which is used to assess whether a “request” is critical or not. The further workflow depends in part on this assessment.

As good as it is to consider the effects of changes to the device at a very early stage, it is important to warn against making this classification only at the beginning and only based on the change request.
Without knowledge of the context of use and knowledge of the internal structure (the system or software architecture), such an assessment – which would ultimately require a hazard and risk analysis – is not possible. Therefore, the product manager or support staff must not be left solely to assess risks. Instead, it is the task of a team consisting of a risk manager, product manager, and system or software architect(s). If the interfaces, e.g., the GUI, are changed, you may need to involve the context experts and doctors.
From a risk management perspective, this change request takes place in the so-called post-production phase and must therefore fulfill the requirements of ISO 14971 (Chapter 9).
c) Change requests and IEC 62304
IEC 62304 also has precise provisions relating to these change requests. On the one hand, it requires the documentation
- as change requests for the developers and
- for trend analysis, e.g., regarding the IEC 62304 problem-solving process.
Then the change requests involve the release to allow something to be changed. IEC 62304 does not refer to change requests here but to the configuration management process.
d) Change requests and ISO 13485 or FDA
Both the FDA and ISO 13485 require corrective and preventive actions. Simply correcting the error is not enough; it is also about preventive measures.
e) Tools for managing change requests
Representatives of tools for managing the application lifecycle include
- JIRA
- TeamFoundationServer
- Polarion with the special add-ons MedPack and RiskPack for IEC 62304 and ISO 14971-compliant documentation
With some ticket systems such as Bugzilla or Mantis, the software lifecycle cannot simply be mapped in accordance with IEC 62304.
4. Software change: What else you should consider
Regardless of whether a software change requires a new submission, you should consider the following:
- Changes to the software – even during development – must follow a process.
- This includes the fact that changes must be released,
- that the software is fully or partially re-tested after the changes (regression tests), and
- that you examine change requests to determine whether further measures such as notification to the authorities or a recall are necessary.
- Your notified bodies may require you to communicate software changes regardless of what the FDA thinks.
