For many manufacturers of medical devices, an FDA inspection is associated with great stress. Many companies are aware of the possible consequences, such as a public warning letter and even criminal prosecution. But they often don’t know how to avoid these consequences without shutting down the entire company for days.
This article shows how
- an FDA inspection works,
- you can prepare your company and
- you should behave during an inspection.
This way, an FDA inspection will be a success for you!
1. FDA inspection: The basics
1.1. Definition
An FDA inspection is a structured procedure in which the FDA examines the conformity of organizations with the requirements of 21 CFR (Code of Federal Regulation), particularly with the quality requirements of 21 CFR part 820. This law is also called QSR, Quality System Regulations.
All organizations that fall under the regulations of 21 CFR must expect FDA inspections. In addition to medical device manufacturers, this also includes pharmaceutical companies and the food industry.
The FDA conducts inspections worldwide, i.e., on-site at the organizations to be inspected.
1.2 Differentiation between inspections and audits
In contrast to an audit, the objective of an inspection is not to identify potential for improvement. It is “only” about identifying violations of the law.
- If successful, an audit leads to a certificate (or its renewal), and if unsuccessful, in the worst case, to the withdrawal of the certificate.
- An unsuccessful FDA inspection, on the other hand, can have more serious consequences (see below).
1.3 Inspection types
Depending on the cause, the FDA distinguishes between different types of inspections.
1.3.1 Pre-Approval Inspections
The FDA conducts a Pre-Approval Inspection for high-risk medical devices that undergo Pre-Market Approval (PMA) as part of the approval process if required. A successful inspection is a requirement for marketing the devices in the USA.
1.3.2 For-cause Inspections
For-cause Inspections always relate to a specific reason. They particularly take place when serious adverse events occurred involving the company’s own device or competitor products or other indications of a violation of the law.
1.3.3 Follow-up Inspections
If the FDA has identified violations during an inspection, it can conduct Follow-up Inspections. In doing so, the authority checks whether the organization has actually implemented the planned corrective actions and remedied the violations.
1.3.4 Routine Inspections
Routine Inspections are the most common type of inspection. The frequency of these inspections depends, among other things, on:
- product type
- risk or class of the device
- result of previous inspections (problems)
- date of the last inspection
- results of inspections/audits by other authorities or bodies (e.g., MDSAP)
During the “COVID years,” the authority initially did not carry out any of these inspections outside the USA. It is now returning to the original pattern.
The FDA has published guidance documents on its inspections. However, some of this information is 25 years old and no longer up to date in all cases.
2. Inspection procedure
From the manufacturer’s point of view, an FDA inspection takes place in several phases (see Fig. 1).
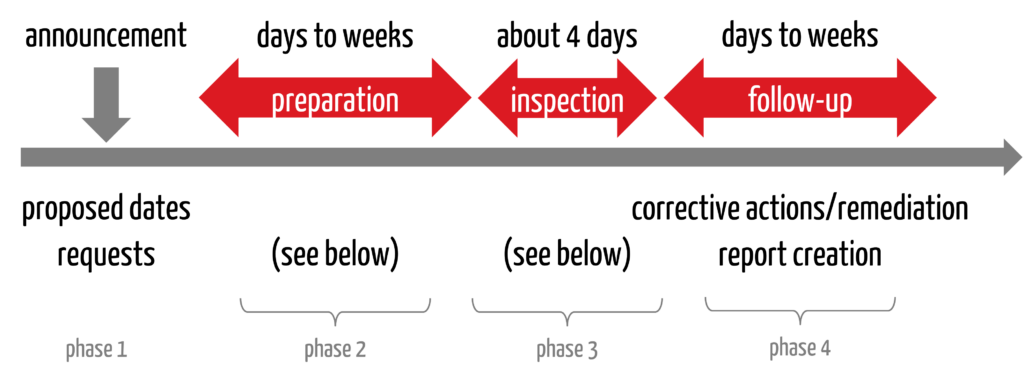
2.1 Phase 1: Announcement and coordination
The procedure begins with the authority getting in touch, usually by email. In this message, the FDA informs you that an inspection will take place. It proposes dates, one of which the companies should accept if possible.
The FDA usually asks companies to send their QM manual to the authority. Not all standard operating procedures, but the manual. This is the FDA’s way of preparing. It will request other information, such as the list of devices currently marketed in the US and whether translators are available during inspections abroad.
Companies based in the USA often only have a few days between the announcement of the inspection and the inspection itself. Outside of the US, companies usually have one to three months.
2.2 Phase 2: Preparation
In this phase, companies should check their quality management system for conformity with the requirements of 21 CFR part 820. An internal audit or a mock inspection (often with external help) can help to resolve possible non-conformities.
Follow the recommendations for preparation in section 4.1 of this article!
2.3 Phase 3: Implementation
The inspection takes place. A routine inspection usually takes four days.
2.3.1 Beginning
Similar to an audit, the inspection begins with an opening meeting. Among other things, the agenda is discussed. The company should briefly introduce itself and its devices in the opening meeting. It may suggest a tour of the company so that the inspector can get an impression of the company or site.
2.3.2 Inspection with the QSIT procedure
The FDA carries out the actual inspection using the QSIT procedure. QSIT stands for Quality System Inspection Technique, a top-down approach. The FDA starts with individual processes and works its way up from there to individual records. The inspector must take a certain number of random samples. The QSIT specifies this number.
2.3.3 Documentation of observations
Deviations, i.e., the inspectional observations, are noted by the inspector on a so-called 483. This is the number of the corresponding FDA form. In addition, the inspector notes discussion items, i.e., points worthy of discussion but which do not necessarily constitute a deviation. These are not documented on the 483 but end up in the final report. This is called the Establishment Inspection Report (EIR).
2.3.4 Closing
At the end of the inspection, the inspector explains the deviations he has found and discussion items with the company.
2.4 Phase 4: Follow-up
After the inspection, the companies must – if necessary – resolve any violations. They should immediately start eliminating initial problems and present a precise action plan to the FDA. The FDA recommends a response to the 483 within 15 working days.
The FDA will finalize the report. The time it takes to finalize and submit the report depends primarily on the potential consequences (see section 3). If no regulatory action is planned, companies can expect the report and the procedure to be finalized within approximately 30 to 50 days.
3. Output of the inspection
The consequences or tasks for a company depend on the inspection output.
3.1 No Action Indicated NAI
No Action Indicated is the ideal case. Then the company has done everything right. There were no findings or violations of the law, and the FDA is not planning any action.
3.2 Voluntary Actions Indicated VAI
The next level is the Voluntary Actions Indicated. In this case, the FDA has found deviations and issued a 483. VAIs are issued for deviations that are below the threshold for serious regulatory action. Nevertheless, the FDA can request a written response from the company and recommend corrective actions. However, these are voluntary.
3.3 Official Action Indicated OAI
Major deviations lead to an Official Action Indicated. There is a real problem that the company needs to address urgently.
In the worst case, these major deviations have further consequences, e.g.:
- FDA warning letter
- import ban
- seizure of the devices
- prosecution measures
Whether this escalation occurs depends on how and how quickly the company reacts and implements any corrective actions.
4. Practical tips for FDA inspections
4.1 Tips for the announcement and preparation phase
Get started immediately and use the time until the inspection to prepare as best possible. Proceed as follows:
4.1.1 Prepare
- Let the management and department heads know immediately. They must be informed and free up resources if necessary.
- Ensure that all persons required for the inspection are available on the inspection days. In addition to the management, this naturally includes the quality management representative, someone from Regulatory Affairs, and the process owners.
- Prepare the necessary technology, e.g., for communication and note taking , and check it before the inspection.
4.1.2 Check conformity
Then, it is time to check the conformity of your QM system if this has not already been done. An internal audit is typically carried out for this purpose. However, internal does not mean that an external person is not allowed to do this. In fact, involving an external person with a high level of competence and an independent view is advisable.
Benefit from our FDA and QM experts to reliably identify non-conformities and eliminate them to the FDA’s satisfaction. Our experts have seen it all. So don’t be afraid to get in touch.
The output of these inspections is a list of deviations and recommendations for improvement.
The top 3 deviations include the CAPA system, complaint handling, and vigilance or adverse event reporting system (MDR reporting). You should, therefore, review these processes particularly closely.
4.1.3 Create and implement a plan to eliminate non-conformities
- Create a CAPA plan and work through it.
- If you discover several significant deviations, a remediation project is due. The objective of this is to eliminate all non-conformities as quickly as possible and in the best possible way before the inspection.
4.1.4 Practice the inspection in a final rehearsal
Another recommendation is to practice the inspection. This includes a final rehearsal. Because everyone in the company needs to know
- how an inspection works,
- how they should behave during the inspection and
- what to say and what not to say so as not to ruin the preparation.
This final rehearsal is also called a mock inspection.
Our experts will train you in the correct behavior and conduct mock inspections. This will ensure that your team behaves professionally during the inspection and does not risk the success of the inspection.
4.2 Behavior during inspections
Tip 1: Answer the inspectors’ questions, but no more.
Tip 2: Don’t play for time. If you are unable to provide the requested evidence, e.g., test reports or training certificates, within a reasonable period of time, you will not only annoy the inspector. You are also provoking a deviation.
Tip 3: Separate the front room, where the inspector sits with your management representative, note taker, and the person responsible for the process currently being inspected from the back room, also known as the war room. There are other people in the back room who have the task of organizing the required documents and requesting and issuing the required records and documents from the company.
Tip 4: If you can define or even implement measures during the inspection, this will be well received.
Tip 5: It may sound banal, but it’s important: treat the inspector in a respectful and friendly manner. Always. Even if you don’t agree with him or her. Don’t become compromising, either.
5. Summary
Keep a cool head when the FDA announces itself for an inspection but act quickly and consistently. After all, you want and need to avoid a negative outcome in order to safeguard your company’s reputation and existence.
The likelihood that your inspection will be successful depends, above all, on the quality of your preparation. In addition to internal audits, this also includes mock inspections, with which you can practice the correct behavior during the FDA inspection.
The FDA experts at the Johner Institute have always been able to avoid an FDA warning letter with their help during inspections. Benefit from this expertise now:
- Ensure QMS compliance via an internal audit and identify any non-conformities
- Create action plans to eliminate non-conformities
- Eliminate non-conformities sustainably by eliminating the root cause
- Practice the correct behavior during inspections in training courses and mock inspections
- Communicate professionally with the FDA before, during, and after inspections
Get in touch to ensure that your FDA inspection is a success with minimal stress and no unnecessary effort and that you have a reason to celebrate afterward. We will discuss your personal roadmap in an initial meeting.
