University institutions in particular regularly publish medical software as open source. This raises doubts as to whether this open-source software counts as a medical device and what regulatory and (product) liability risks are involved.
This article provides a quick overview.
- The legal and financial risks depend on whether the open source software counts as a medical device and whether it has been placed on the market.
- The regulatory requirements do not depend on whether software is placed on the market as open source.
- Developers of open source software should and can avoid the unintentional placing on the market of a medical device and the associated liability risks.
- Even in the case of “open source medical devices,” not only the product but also the manufacturer/marketer must fulfill many legal obligations.
1. Role of open source software in medical devices
Open source software can be both the medical device and a part of it. The following constellations are possible:
- The open-source software is a component developed specifically for the medical device (e.g., an AI model with associated API).
- The open-source software is an off-the-shelf component used in the medical device, such as a library.
- The open source software is the medical device itself.
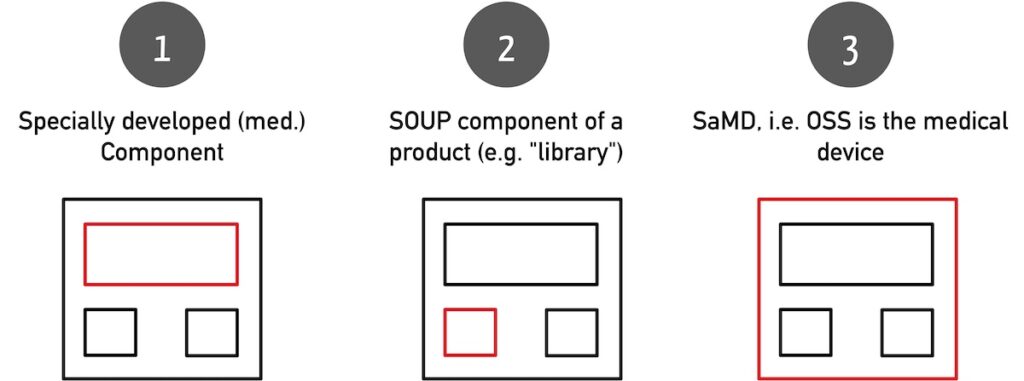
Please note the differences and similarities between off-the-shelf software (OTSS) and SOUP.
2. Legal classification
Liability arises in particular when a medical device is placed on the market. Therefore, two questions must be clarified:
- When does a product count as a medical device?
- When is a product considered to have been placed on the market?
a. When does a product count as a medical device?
It is not the functionality of a product that determines its qualification (decision as to whether it is a medical device or not), but its intended purpose.
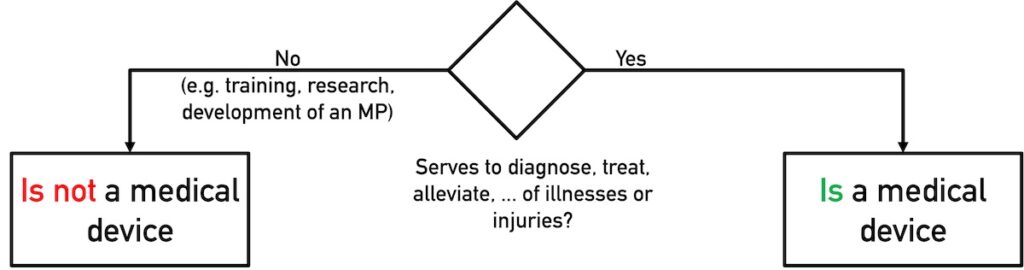
Consequently, open source software developers avoid their work results being classified as medical devices in the following intended uses, for example:
- The product is used for research or the documentation of research results.
- The software is intended/may be used to develop medical devices.
- The open source software is used for training purposes.
It is particularly helpful to explicitly note if/that the software is not a medical device and is not intended for the diagnosis, alleviation, treatment, or prediction of diseases and injuries.
A (regrettable) exception to the rule that the intended purpose and not the functionality determines the qualification can be found in MDCG Guideline 2019-11. According to this, software that is only used for the (unchanged) transmission, storage, and display of data does not count as a medical device.
The definition of what constitutes a medical device varies in different jurisdictions. For example, according to the US FD&C, many software applications are not subject to FDA enforcement.
b. When is a device considered to be placed on the market?
The MDR defines what constitutes placing on the market in Article 2 (sections 27 and 28):
any initial supply, whether in return for payment or free of charge, of a product […] for distribution, consumption, or use on the Union market in the course of a commercial activity;
Article 2 MDR, sections 27 and 28
The consumption or use of a medical device in a doctor’s office or hospital counts as a commercial activity, even if this activity is not profit-oriented.
According to the definition, it does not matter whether the supply is for remuneration or free of charge, as is often the case with open source software.
If the open source software is only intended for use within the healthcare facility where it was developed, in-house production in accordance with MDR/IVDR Article 5, Section 5 may be considered.
3. Legal obligations
a. Medical device law
If the open source software is considered a medical device that is or will be placed on the market, its manufacturers must comply with all regulatory requirements for medical devices.
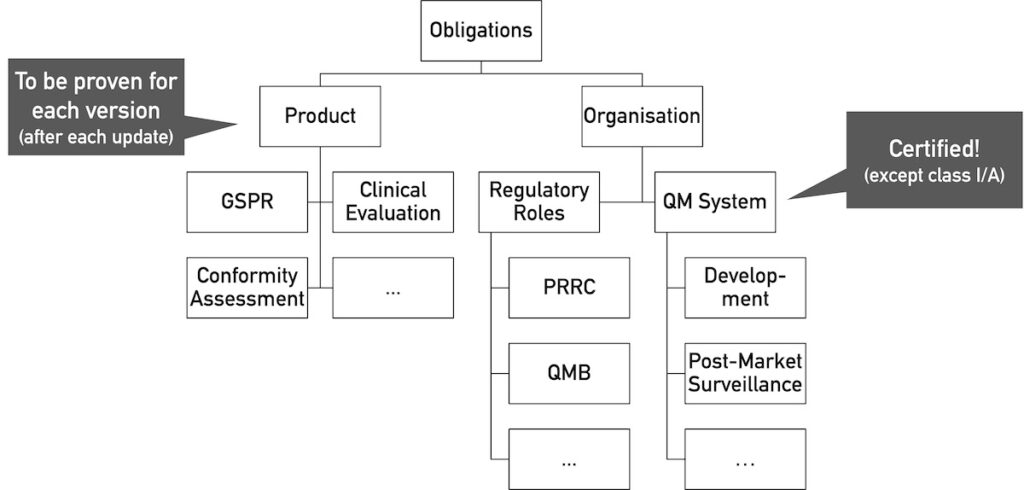
These requirements apply to both the medical device and the organization that places the medical device on the market.
The most relevant general safety and performance requirements (GSPR) for software concern:
- Risk management (according to ISO 14971)
- Software development processes (according to IEC 62304)
- Usability engineering (according to IEC 62366-1)
- IT security (according to IEC 81001-5-1)
In addition, a quality management system in accordance with ISO 13485 or Annex IX of the MDR/IVDR is required. This also includes a post-market surveillance system.
Unfortunately, many German authorities insist on classification in class IIa or higher, regardless of the legal requirements in Annex VIII of the MDR (especially Rule 11).
However, most legal obligations, such as the GSPR, are largely independent of the class of the software.
This article describes the steps involved in placing a medical device on the market.
b. Additional legal requirements
In addition to and independently of medical device law, manufacturers may have to comply with other “horizontal” regulations, for example:
- AI Act
- EU Data Act
- EU Health Data Space
- IT security requirements such as NDIS2
- Product Liability Act/Regulation/Directive
The applicability and relevant requirements of these regulations depend on the role played by open source developers.
The AI Act does not impose any requirements on open source AI systems.
4. Evaluation and application examples
a. Advantages of open source medical software
The developers of open source medical devices argue that it offers the following advantages:
- Open source promotes innovation and brings innovation quickly to the patient’s bedside.
- The development results are available to everyone, regardless of their financial means.
- Transparency and the community approach help to quickly find and eliminate errors, which serves patient safety.
- There is no conflict of interest, as is the case with manufacturer-funded clinical investigations.
b. Challenges with open-source medical devices
The development of open source medical devices must overcome specific challenges:
- It requires an organization with a (certified) quality management system and all regulatory roles such as the quality management respresentative and the PRRC.
- Competencies must also meet regulatory requirements outside of development, for example in quality and risk management, usability engineering, and regulatory affairs.
- The high post-market costs must be incurred and financed over many years, at least as long as a product is still on the market.
- Since there is no seller-buyer relationship, communication with customers/users is more difficult, which makes risk management and post-market surveillance, among other things, more challenging.
- Staff turnover, for example among doctoral students, makes it challenging to secure skills on a permanent basis.
- The control (creation, review, approval) of artifacts such as documents and source code requires special attention in a community that does not correspond to an organization.
The diabetes dosing system from Tidepool is based on open-source software. The company has described its regulatory journey here.
5. FAQ
a. How can I avoid being sued as an open source developer?
The risk of being sued cannot be ruled out, but the probability can be lowered. The risk is greater
- the more obvious a possible violation of the law is,
- the higher the economic relevance of the product (e.g., for competitors), and
- the more financially powerful the defendant.
It is therefore helpful to formulate the intended purpose and claims in a very consistent and transparent manner and to research and comply with regulatory requirements.
The Johner Institute also supports developers of open source products with their regulatory strategy. On the one hand, with the aim of either avoiding or achieving qualification as a medical device, and on the other hand, with the aim of achieving regulatory security and compliance.
If you are interested, simply get in touch using the contact form.
b. Do I have to affix a CE mark? If so, where?
With the CE marking, the distributor declares conformity with applicable EU directives or regulations. This marking is only permitted and required if the product falls within the scope of one of these directives or regulations and the CE marking is required there.
Please also refer to the technical article with details on CE marking and the affixing of the CE mark on software.
c. What do I need to bear in mind when using GitHub?
IT systems for version and configuration management and for operating CI/CD pipelines are considered computerized systems within the scope of the QM system and must therefore undergo computerized systems validation.
These systems must ensure the document control required by law and standards, which is more a question of correct configuration than of the features of these systems.
d. How do you document an open-source SaMD for FDA approval?
Open-source SaMD is subject to the same regulatory requirements as all other SaMD. These include:
- Technical documentation comparable to MDR
- Cybersecurity file as part of the technical documentation
- Quality management according to 21 CFR part 820
- FDA approval, for example, as part of a 510(k) or de novo approval.
e. Who can help me?
The (software) experts at the Johner Institute can help with all questions relating to documentation, approval, and establishing a management system. They can also assist with the required tests, such as usability tests, pentests, and clinical investigations.
6. Conclusion and summary
Many university institutions underestimate the effort required for the legally compliant development, marketing, and monitoring of medical devices on the market. Professional software engineering is a necessary but by no means sufficient prerequisite.
It can be helpful to separate research and development and not to rule out commercialization altogether, especially since this does not exclude the open-source concept. After all, doing good for patients not only costs time, but unfortunately also money.


