The Medical Device Coordination Group (MDCG) has drafted a guidance document that describes how manufacturers should place their class 1 medical devices on the market in order to comply with the MDR.
The title of the document is “Guidance Notes for Manufacturers of Class I Medical Devices”. This article provides a summary of the document and gives useful tips for manufacturers of class 1 medical devices.
1. What characterizes class 1 medical devices
a) Grouping medical devices into classes
The Medical Device Regulation (MDR), just like the Medical Device Directive (MDD), splits medical devices into classes 1, 2a, 2b, and 3. The classes are often written using Roman numerals (class I, IIa, IIb, and III).
The classification rules assign devices with higher risks to the higher classes.
Class I medical devices include surgical drapes and many surgical instruments such as tweezers, latex gloves, masks, wheelchairs (without a motor), stethoscopes, bandages, or spectacles.
b) Effect of classification
The classes do not have any(!) effect on the general safety and performance requirements that manufacturers have to demonstrate compliance with. Instead, the classes determine which conformity assessment procedure is required to demonstrate conformity with these same safety and performance requirements.
A notified body does not have to be involved in the conformity assessment procedure for class 1 medical devices. In addition, for class 1 medical devices, the MDR does not insist on the certification of the quality management system by a notified body. Both these things save time and money.
Class 1s, 1r, and 1m medical devices are exceptions to this rule:
- 1s: Devices that are placed on the market in sterile condition
- 1r: Reusable surgical instruments (r stands for “reusable”)
- 1m: Devices with a measuring function
For these “class 1* devices”, manufacturers must involve notified bodies in the conformity assessment.
Read more about the classification of medical devices and the conformity assessment procedure.
Please also note the information on class I software.
2. How class 1 medical devices are “approved”
The MDCG describes eight steps that manufacturers should go through when placing their devices on the market.
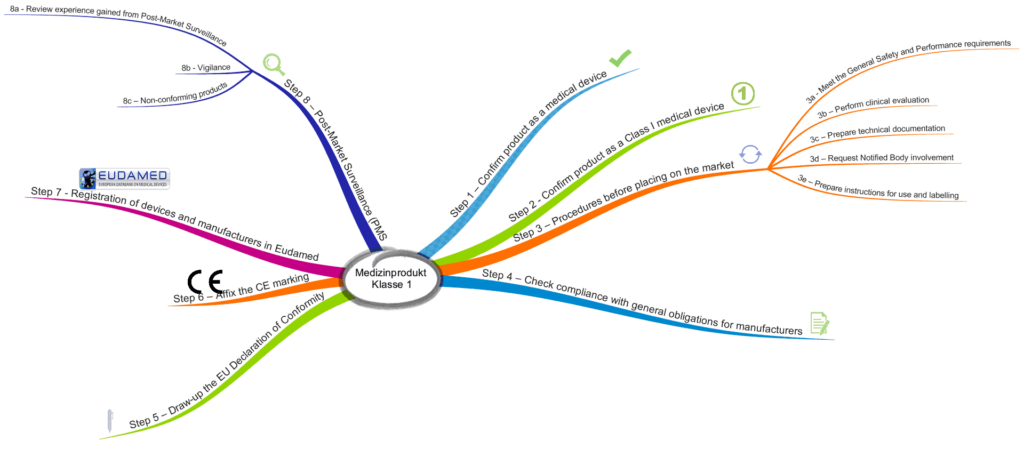
Step 1: Check and confirm that the device is a medical device
The MDR has taken the definition of the term “medical device” almost unchanged from the MDD. It is, therefore, unlikely that a device that was previously a medical device will now be outside the scope of the MDR.
Step 2: Confirm that the device is a class 1 medical device
This step is essential because the MDR has changed the classification rules. For example, almost all standalone software is no longer (!) in this lowest class.
The MDCG guidance document that this article is discussing only applies to class 1 medical devices, although most of the requirements apply to all medical devices.
Step 3a: Ensure that the general safety and performance requirements have been met
Obviously, the general safety and performance requirements established in Annex I must be complied with. However, the MDCG reminds us that other regulations, such as the Machinery Directive, must also be complied with.
It emphasizes the importance of risk management and reminds us that compliance with the requirements can also be demonstrated through the application of harmonized standards and common specifications.
Step 3b: Perform clinical evaluation
The MDCG also reminds us how important the clinical evaluation is to the MDR and that the MDR insists that manufacturers:
- analyze and compare alternative treatments
- take post-market data into account
- demonstrate the acceptability of the benefit-risk ratio based on the clinical data
- perform clinical investigations if they do not have sufficient clinical data
Step 3c: Prepare technical documentation
According to this step, manufacturers of class 1 medical devices must prepare the technical documentation in accordance with annexes II and III. Naturally, this applies to all medical devices.
The MDCG document looks at some of the changes introduced by the MDR compared to the MDD:
- justification of the classification
- Basic UDI-DI
- reference to predecessor devices and similar devices
- reference to applied and valid standards and common specifications
- description of the post-market surveillance system
Step 3d: Request notified body involvement
As mentioned above, manufacturers of class 1* medical devices must involve a notified body accredited for the corresponding device classes:
- “Devices in sterile condition”: Code MDS 1005
- “Reusable surgical instruments”: Code MDS 1006
- “Devices with a measuring function”: Code MDS 1010
Manufacturers can use the NANDO database to see which notified bodies are accredited for each code.
Step 3e: Prepare instructions and labeling
Somewhat surprisingly, it is only in step 3e that the MDCG looks at the instructions for use and labeling. Ultimately, Annex I determines their content. The instructions for use are also required for the clinical evaluation.
The MDCG mentions that no instructions for use are required for class 1 medical devices if safe use is guaranteed. The document also looks at language requirements (without giving a list of required languages) and distributors’ obligations to provide these accompanying materials in these languages.
Symbols would be provided by harmonized standards and common specifications, and the label has to make clear that the device is a medical device.
Step 4: Check compliance with general obligations for manufacturers
This fourth step is less about the device and more about the manufacturer, in particular the manufacturer’s obligation to set up a QM system and take out insurance.
The MDCG does not look at the new obligation to have a competent person according to Article 15 at this point. It actually does this in the introduction.
Step 5: Create the EU declaration of conformity
Less surprising is the requirement for the manufacturer to declare conformity with the MDR and other EU regulations and to translate this declaration of conformity into the national language of the country the device is placed on the market in.
Step 6: Affix the CE marking
This step is also obvious: Manufacturers of class 1 medical devices must also affix the CE marking. In the case of class 1* medical device, the CE marking must be accompanied by the identification number of the relevant notified body.
Step 7: Registration of devices and manufacturers in EUDAMED
However, the seventh step is new in its current form. Manufacturers must register themselves in EUDAMED and are assigned an “SRN”. The also applies, incidentally, for EU representatives and importers.
It is then the manufacturer’s responsibility to register a medical device. They then get assigned a UDI-DI and a Basic UDI-DI.
Until EUDAMED is fully functional, manufacturers and EU representatives must notify the competent authority (in Germany, BfArM) and register the device.
Step 8a: Collect and evaluate post-market data
The MDR requires manufacturers to have a post-market surveillance system that is part of the QM system. For class 1 medical devices, manufacturers must create a “Post-Market Surveillance report”. The requirements for this report are not as stringent as they are for the “Periodic Safety Update Report” (PSUR).
Step 8b: Vigilance system
The MDCG gives a reminder that the MDR also requires manufacturers of class 1 devices to report “Field Safety Corrective Actions” (FCSA). Until EUDAMED is in operation, these notifications are sent to the national authorities (in Germany BfArM and in Switzerland SwissMedic). These reports will be subsequently recorded in EUDAMED.
The MDCG document describes, relatively comprehensively, the obligations in the event of FCSAs as they are (still) currently defined by the German Medical Device Safety Plan Ordinance.
Step 8c: Handling non-conforming devices
The last step concerns the procedure in case of non-conforming devices. Apart from the reporting obligations mentioned above (step 8b), manufacturers must make appropriate corrections and/or take appropriate corrective action.
Our video training and dozens of templates give you everything you need for fast, MDR/IVDR-compliant approval. Individual onboarding and a customized project plan make it easy to stay on track and complete all the necessary steps.
3. What authorities check class 1 devices for
In contrast to higher-class devices, monitoring class 1 devices and their manufacturers is primarily the authorities’ responsibility. These authorities, e.g., trade supervisory offices and regional councils, are increasingly fulfilling this monitoring obligation. They rely on checklists, such as the following exemplary list. Please note, however, that not all checklists look the same.
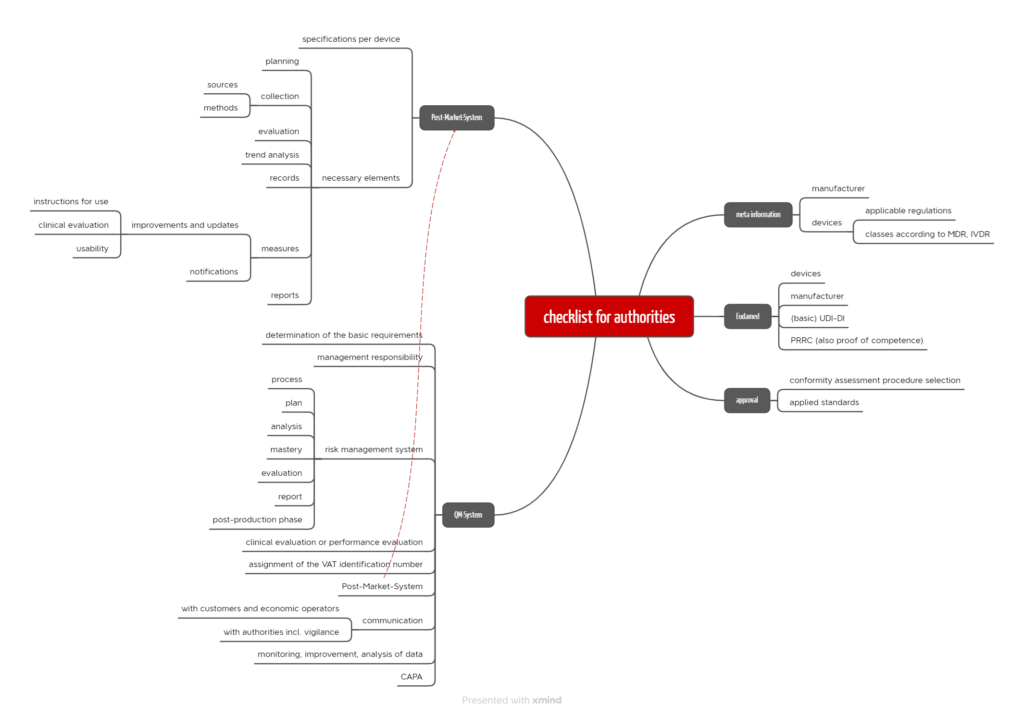
Authorities checklist-1 Download
As expected, the authorities’ checklists are strongly based on the MDR and IVDR. More surprisingly, this checklist focuses on the requirements for the risk management system, post-market surveillance, and vigilance. Here, the authorities address almost every sub-sentence of the regulatory requirements.
4. Conclusion
The 20-page document “Guidance Notes for Manufacturers of Class I Medical Devices” should – as the name suggests – is intended to act as a guideline for manufacturers of class 1 medical devices.
The document meets this target to some extent. However, it should be pointed out that the requirements it contains are not complete and not fundamentally specific to class 1 medical devices.
The Johner Institute helps class 1 manufacturers to quickly and easily comply with regulatory requirements and thus be perfectly prepared for inspections by authorities. Contact us, e.g., via our web form.
Change history
- 2025-03-28: Examples added to section 1.a)
- 2023-03-03: Notes on articles on class I software added
- 2021-09-26: Headings numbered and chapter 3 added

It is mentioned ‘’The Medical Device Coordination Group (MDCG) has drafted a guidance document that describes how manufacturers should place their class 1 medical devices on the market in order to comply with the MDR.
The title of the document is “Guidance Notes for Manufacturers of Class I Medical Devices”. This article provides a summary of the document and gives useful tips for manufacturers of class 1 medical devices.‘’ at the beginning of this article which was issued on March 28, 2025 . That means “Guidance Notes for Manufacturers of Class I Medical Devices” was not published at least before March 28, 2025. However, “Guidance Notes for Manufacturers of Class I Medical Devices” has been published in December 2019, see the link below.
https://health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en
So the content at the beginning of this article is quite confusing. It would be grate if this content could be checked again and modifty the parts which may cause misundersting of readers.
Dear Mr. Chen,
thank you for the comment. We will check this and make any necessary changes.
Best regards