Laws and standards require organizations to prepare a risk management report. Notified bodies and authorities examine these reports intensively because risk management is a key regulatory requirement.
Therefore, it is important (not only) for manufacturers to prepare precise, complete, and correct risk management reports. This article provides assistance in this regard.
1. What is a risk management report
a) Risk management report from the perspective of ISO 14971
The risk management report is a part of the risk management file. According to Chapter 9 of ISO 14971, it must contain the “outputs of the review” of the risk management activities, in particular:
- Implementation of the risk management plan
- Justifiability of the overall residual risk
- Information for the post-production phase
Chapter 4 of this article provides further details on the contents of this risk management report.
The risk management report is often a stand-alone document. However, this is neither required by regulation nor is it the most pragmatic approach in any case. More on this in chapter 4 as well.
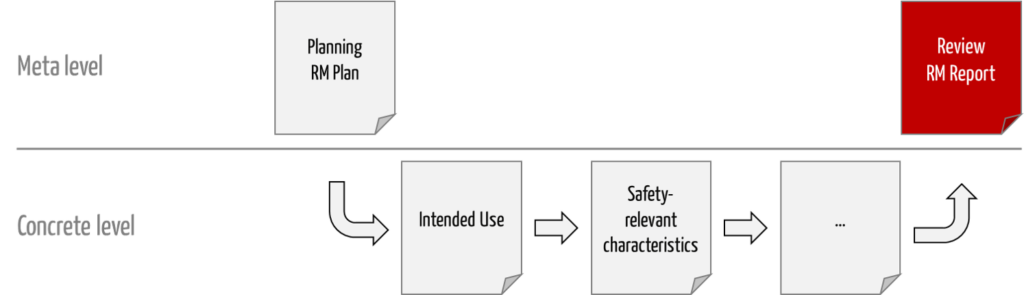
b) Delimitations
Reports on the effectiveness of the risk management process
In Chapter 4.2, ISO 14971 requires an organization’s top management to review the suitability [and effectiveness] of the risk management process at planned intervals. Organizations must document the outputs of this review.
However, these reports on the effectiveness of the risk management process should be distinct from risk management reports. The former is at the management level (e.g., as part of the management review, ISO 13485:2016, Chapter 5.6), and the latter at the product level.
Report on risks from the post-production phase
In this presentation, FDA describes the “Risk Management Report” as a document that contains an assessment of post-production phase information relevant to risk. Such a narrowing of focus is unusual.
Evaluation of the company risk
ISO 9001 also sets requirements for dealing with risks. The organizations must also recognize and deal with these risks (Chapters 0.1 and 0.3.2) or consider them in their quality system (Chapters 4.4 and 6.1).
Reports on these analyses and measures are also not a “risk management report”. These contents would rather be part of a management report.
2. Why you need a risk management report
a) Regulatory requirements
The regulatory requirements are the most important reason for many organizations to prepare a risk management report.
- The MDR and IVDR do not require a risk management report in their respective Annexes I and II. However, they require the content typically found in such a report.
- ISO 14971 explicitly insists on a risk management report (Chapter 9).
- Similar to the EU regulations, the FDA does not insist on a report. But it, too, requires that manufacturers compile the information it contains.
- ISO 13485 requires manufacturers to document the risk management process and risk management activities. The latter would be part of a risk management report.
The FDA’s considerations in the guidance document Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement Decisions are interesting.
The risk management report helps to implement the PDCA concept. This is because it contains the check whether the risk management activities are following the plan.
b) “Executive summary” for authorities and notified bodies
Regardless of regulatory requirements, a risk management report is an important document for authorities and notified bodies. They want to benefit from it as an “executive summary” for risk management because there is usually not enough time to evaluate the complete risk management file.
c) Safety and security
Using the risk management report, manufacturers can ensure that they have done everything possible to
- the safety and security of the devices,
- the safety of patients and, last but not least,
- the security of their own company.
3. Who needs a risk management report
Risk management reports, in the sense of this article, are required (at least) by all organizations that must comply with the requirements of ISO 14971. This also concerns the organizations that declare conformity with ISO 13485 because ISO 13485 requires risk management and recommends one in accordance with ISO 14971.
These organizations include:
- Medical device manufacturers
- Manufacturers of medical device accessories
- Their suppliers, if applicable
- Importers, distributors, and others covered by Article 16 of the MDR
- Organizations that reprocess single-use devices (see Article 17 of MDR)
These organizations require a risk management report for each medical device.
The MDR allows manufacturers to create the technical documentation (TD) for all devices in a base UDI-DI. The risk management files are part of this TD.
4. What a risk management report should contain
a) The academically correct approach
If one restricts oneself to the requirements of ISO 14971, then the risk management report contains only the outputs of the inspection as to whether
- risk management has been carried out as specified in the plan,
- the overall residual risk is acceptable, and
- appropriate methods are in place to collect and review information in the production and post-production phases.
This report contains only the “judgments” and justifications for them. That is, the ISO 14971 risk management report is a meta-level document (see Fig. 1).
Manufacturers can use a spreadsheet to demonstrate that the plan has been implemented:
| Activity according to plan | Activity carried out | Objective evidence | Comment |
|---|---|---|---|
| Description of the activity | [ ] yes [ ] no | Reference to documents and records | e.g., notes on why the plan was deviated from |
ISO 24971 contains little additional information on the risk management report. It seems to follow the approach of limiting this report to the three pieces of information mentioned above.
b) The pragmatic approach
In fact, the risk management report is often expected to provide an “executive summary” of the overall risk management process. Therefore, organizations may supplement the report with additional information:
- Summary of risk policy and risk acceptance criteria
- List of key hazards/risks
- List of key risk control measures
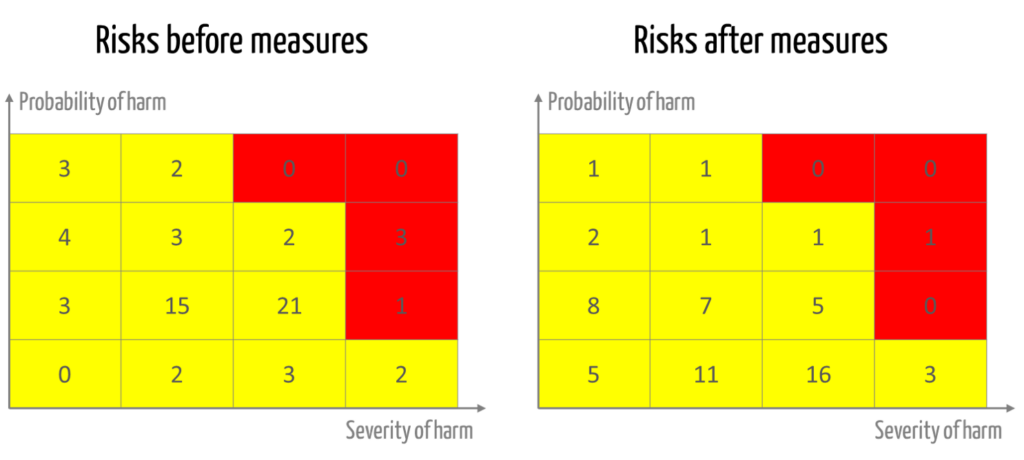
- An overview of the risks before and after the measures (see Fig. 2)
In some cases, manufacturers go so far as to prepare only three documents:
- Risk management plan
- Risk table
- Risk management report (contains everything else)
A risk assessment matrix can be used to demonstrate how the risks have changed as a result of the measures.
See our tips for creating a risk assessment matrix and risk acceptance criteria.
5. What to look for in the risk management report
a) Ensuring the competence of the responsible persons
Manufacturers must define and review the competencies of the persons involved in risk management and document both. In Chapter 7.3.2, ISO 13485 requires these specifications for the respective development project.
This makes sense when writing the risk management plan. For example, for the benefit-risk evaluation, the responsible persons must know and evaluate the following:
- Product-specific risks
- Concrete measures to reduce the risks
- State-of-the-art (thus also alternative devices and procedures)
b) Establish criteria also for the overall risk-benefit assessment
The manufacturers document their risk acceptance criteria in the form of a risk acceptance matrix (see Fig. 2). This matrix is well suited for deciding on the justifiability of individual risks. However, it is not suitable to the same extent for justifying why
- risks that remain in the “red zone” even after measures are taken are nevertheless accepted in view of the overall benefit-risk ratio,
- the risks remaining in the yellow and green ranges are acceptable in their aggregate.
Manufacturers should define the criteria for these justifications.
c) Update reports as needed
A reviewed and approved version of the risk management report before the first placing on the market of the device is mandatory. However, this does not mean that this report will no longer be changed. ISO 24971 writes in this regard:
There can be a need to revise or update the risk management report if new information becomes available, for example during the production and post-production phases.
ISO 24971, Chapter 9
6. Summary and conclusion
The risk management report is an indispensable part of any technical documentation.
ISO 14971 prescribes the “minimum contents” of this report.
Manufacturers can divide the contents into different documents:
- They can limit the risk management report to this content and write an additional “executive summary” on risk management for authorities and notified bodies, or
- they can supplement the risk management report with the content that the authorities and notified bodies expect in an “executive summary.”
Many manufacturers find it most difficult to justify why the remaining residual risk is acceptable in view of the benefits and the state of the art.
The Johner Institute supports medical device manufacturers in structuring and creating risk management files, formulating valid benefit-risk arguments, and evaluating the acceptance of risks and the effectiveness of measures. Feel free to get in touch, e.g., via our contact form.
Change history
- 2023-03-20: Article completely revised
- 2016-04-26: First version created