Legal Requirements and Practical Implementation under MDR/IVDR
EU Authorized Representatives are not only subject to the legal requirements of the MDR and IVDR. They are also liable for violations of medical device law.
This expert article provides practical answers to all regulatory questions relating to the EU-REP with specific recommendations for action from the leading consulting firm for medical device regulation.
It is aimed at non-EU medical device manufacturers as well as regulatory affairs managers, quality managers, PRRCs, and managing directors, but also at EU authorized representatives.
- An EU authorized representative is mandatory for all non-EU manufacturers and is equally liable with the manufacturer.
- The EU REP performs a formal document review but does not assume any manufacturer obligations such as risk management or QM system.
- The appointment is made by written mandate for at least one generic product group and must be registered in EUDAMED.
- A separate PRRC is mandatory and must not be identical to the manufacturer’s PRRC.
- Subsidiaries can be EU-REPs, but they must be legally independent and have their own PRRC.
1. What is an EU representative and when do I need one?
An EU representative is any natural or legal person established in the Union who has been authorized in writing by a manufacturer established outside the Union to perform certain tasks on their behalf (MDR Art. 2 No. 32).
The EU REP…
- is mandatory for all non-EU manufacturers (without exception).
- must be appointed before the first placing on the market.
- applies equally to MDR and IVDR.
- can be appointed per generic product group.
- may already provide support before market launch.
The EU Commission has clarified in a Notice to Stakeholders that EU manufacturers do not need an authorized representative in Turkey and, conversely, Turkish manufacturers do not need an authorized representative in the EU (see technical article: Medical devices in Turkey: Regulatory requirements).
2. The most important tasks and responsibilities of an EU REP
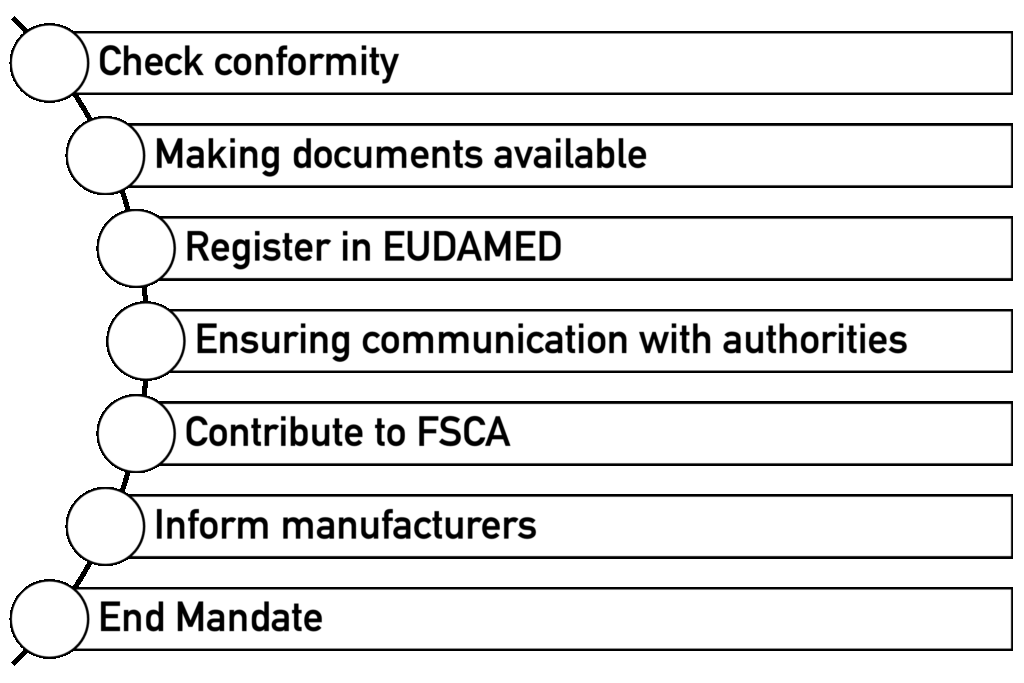
The EU REP has clearly defined mandatory tasks according to MDR/IVDR Art. 11 (3), which must be specified in the mandate:
- Verify conformity: Formal verification that technical documentation has been prepared in accordance with the manufacturer’s QM requirements and that declarations of conformity and certificates are available
- Making documents available: 10–15 years of retention obligation and provision to authorities
- EUDAMED registration: Obtaining own SRN and checking and approving manufacturer registration
- Ensuring communication with authorities: Providing documents in the respective official language and forwarding authority inquiries to manufacturers
- Participate in safety corrective actions in the field (FSCA): Actively cooperate in corrective actions
- Inform the manufacturer: Immediately inform the manufacturer of any incidents
- Terminate the mandate: If the manufacturer violates its obligations, the EU-REP must be able to terminate the cooperation and inform the authority.
3. Liability and legal risks: What is the EU-REP responsible for?
The EU-REP is liable for defective products on the same basis as the manufacturer (Art. 11 (5) MDR/IVDR). This means:
- Equal product liability with the manufacturer
- Liability insurance strongly recommended
- Contractual liability provisions possible
- No assumption of manufacturer obligations under Art. 10
- Support with manufacturer obligations permitted
Ensure that responsibilities are clearly defined in the mandate!
Please also refer to the technical article on product liability for medical device manufacturers.
4. EU REP selection: Importer, subsidiary, or external service provider?
The choice of EU REP depends on the company structure, available resources, and regulatory requirements. There are several options:
| Other role | Option to assume EU REP? |
| Importer | Legally possible, but note role conflicts |
| Subsidiary | Only possible with legal independence and own PRRC |
| PRRC constellation | Must not be identical to manufacturer PRRC (MDCG 2022-16) |
| External service provider | Recommended if internal resources are lacking |
| Timing | Ideally, appoint before conformity assessment |
| Letterbox company | Not sufficient, as physical presence is required |
Pay close attention to the obligations of PRRCs.
Selection criteria for external EU representatives
- Regulatory competence
- Availability in own time zone and in EU time zone
- Experience in the role of EU REP
5. Regulatory inquiries and market surveillance: processes and obligations
The EU REP is the central point of contact for authorities with clear process specifications. Its obligations include:
- Checking documents for completeness in accordance with the manufacturer’s QM specifications
- Providing documents in the official language of the respective member state
- Forwarding product sample requests and executing them
- Active cooperation in market surveillance measures
- Immediate notification of the manufacturer in the event of incidents
- Mandate submission at the request of authorities
There is no obligation for the EU-REP to have a certified QM system. However, authorities or notified bodies may audit the EU-REP to verify that it is fulfilling its obligations.
By checking the documents, the EU representative ensures that the manufacturer has drawn up the EU declaration of conformity and the technical documentation and, if applicable, that a conformity assessment procedure has been carried out (Art. 11, para. 3 lit. a). This check also minimizes his own liability risks.
6. Practical implementation: From designation to labeling
The correct implementation of the EU REP requires a systematic approach, from contract drafting to product labeling. The necessary steps include:
- Agree on a mandate: In writing, signed by both parties, clear definition of tasks; also make a binding agreement on the change of EU REP
- Perform EUDAMED registration: SRN assignment and role confirmation
- Add labeling: “EC | REP” symbol in accordance with DIN EN ISO 15223-1:2022-02 on packaging
- Add EU declaration of conformity: EU REP information mandatory
- Optionally add instructions for use: Information recommended, not mandatory
The EU-REP information should be placed in accordance with DIN EN ISO 15223-1:2022-02 together with the corresponding symbol. The standard is available from DIN Media and as EVS-EN ISO 15223-1:2021 here at a reasonable price. According to this, the EU-REP must be displayed with the symbol “EC | REP”.

A new draft of DIN EN ISO 15223-1/A1:2024-06 is already available, which proposes a change of the symbol from EC-REP to EU-REP.
7. International differences: EU-REP vs. UK-RP, CH-REP, and US agent
Each jurisdiction has specific requirements for representatives—with relevant differences. Examples of these differences are:
- EU-REP: One authorized representative per generic product group possible; requires maintenance of a PRRC
- UK-RP: Only one “UK Responsible Person” allowed for all products
- CH-REP: As in the EU; additionally requires representation PRRC
- US agent: Has different tasks and liability regulations
Many service providers, such as the Johner Institute, take on this role for several markets. This minimizes the effort involved in drawing up contracts, checking documents, and communicating.
Please also refer to the technical articles on the regulatory system in the UK and in Switzerland.
8. Summary & conclusion
The EU authorized representative is much more than a regulatory formality—it is an integral part of the market access strategy for non-EU manufacturers. With equal product liability, extensive documentation requirements, and central communication with authorities, the EU REP bears considerable responsibility. Careful selection, taking into account PRRC requirements, liability risks, and operational capabilities, is crucial for success. As an experienced partner, the Johner Institute supports you in navigating these complex regulatory requirements.
Johner Medical GmbH assumes the role of your EU REP—with years of expertise, its own PRRC, and comprehensive liability insurance.
Contact us for a non-binding consultation.
