1. Definitions
Medical software includes all software used for healthcare, particularly for medical devices or medical devices (embedded software), and software that is itself a medical device (standalone software).
IEC/CD1 82304-1 (Health Software – Part 1: General requirements for product safety) distinguishes between the following terms:
- HEALTH SOFTWARE
Software intended to be used specifically for maintaining or improving health of individual persons, or the delivery of care
- MEDICAL SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device or intended to be a SOFTWARE MEDICAL DEVICE
- SOFTWARE MEDICAL DEVICE
Software intended to be a medical device in its own right
- MEDICAL DEVICE SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device
This clarifies that medical software can be a medical device but does not have to be.
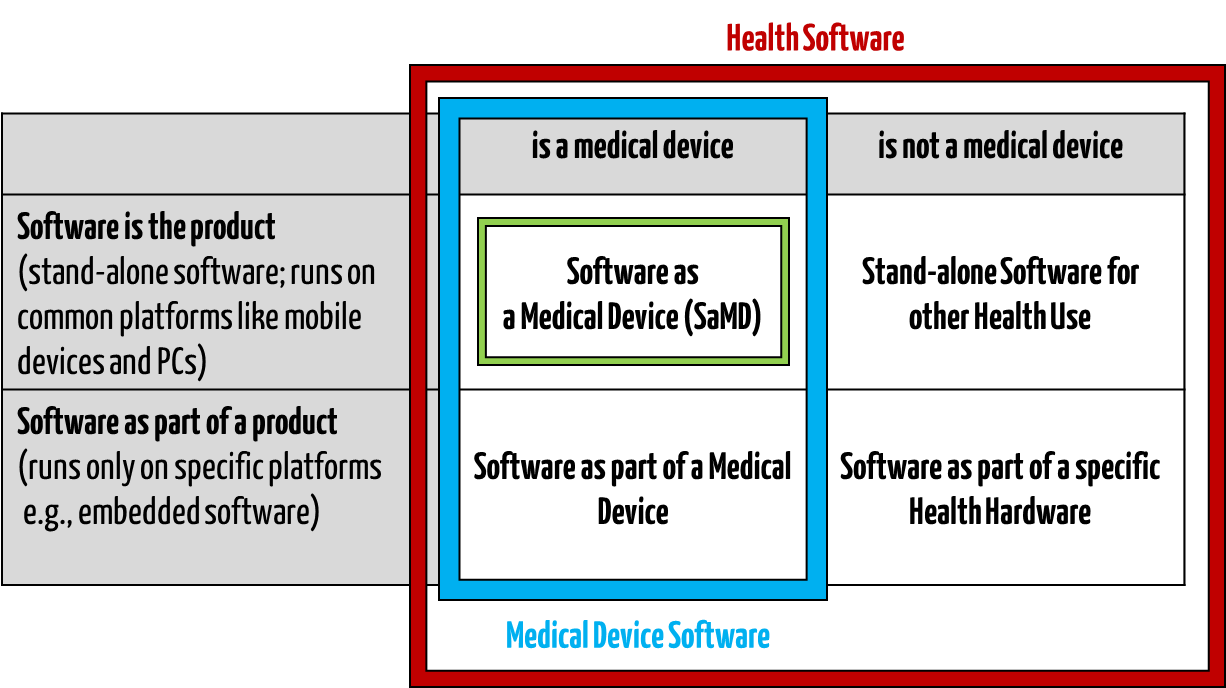
Fig. 1: Medical software includes medical device software and software as a medical device (click to enlarge).
2. Regulatory requirements
a) Medical software – a medical device?
The question often arises as to when medical software meets the definition of a medical device. You can find a further discussion on this topic in the article on the classification of software as a medical device and in the article on the qualification and classification of IVD medical device software.
b) Regulations, laws, standards
Software that is a medical device or part of a medical device must meet the regulatory requirements:
- In Europe, the medical device regulations (MDR, IVDR) are relevant. However, these only contain relatively general regulations for software, which this article presents.
- IEC 62304 defines the life cycle processes for medical device software.
- IEC 82304-1 applies to all “health software”. IEC 82304-1 also requires conformity with the requirements of IEC 62304.
- There are also MDCG guidelines, e.g., MDCG 2019-11 and MDCG 2023-4.
- The FDA sets out specific requirements in its guidance documents, including specific requirements for medical software. It also answered many questions specifically about software as a medical device in this FAQ.
3. Support for medical device manufacturers
Benefit from the support of the Johner Institute:
Contact us right away so that we can discuss the next steps. This will ensure that the “approval” is a success and that your software or devices are quickly launched on the market.
MDCG published guideline MDCG 2023-4 in October 2023 entitled “Medical Device Software (MDSW) – Hardware combinations – Guidance on MDSW intended to work in combination with hardware or hardware components.”
Both the FDA and IEC 62304 recognize software developed by third parties. They refer to Off-the-Shelf Software (OTS) and Software Of Unknown Provenance (SOUP). What is the difference between OTS and SOUP? What do they have in common? What legal requirements do they have to meet? This article provides answers.
Details
For manufacturers, the answer to whether and when clinical studies are necessary when using artificial intelligence in medical devices is relevant. After all, the duration and cost of bringing these devices to market depend on this. The good news in advance: there are cases where manufacturers can avoid clinical studies for devices with AI. This…
Details
The term “medical device PC” is not clearly defined. However, most people understand a medical device PC to be Depending on the constellation, manufacturers must fulfill different regulatory requirements. These are presented in this article.
Details
IEC 80001-1 has the long title “Application of risk management for IT-networks containing medical devices – Part 1: Tasks, responsibilities and activities“. This article reveals what the standard requires and why manufacturers should also consider it.
Details
Many medical device manufacturers create a “software FMEA.” However, there is no uniform understanding of what a software FMEA is. This article provides clarity and tips on how to avoid the most common mistakes.
Details
The V-model is a development process model that was originally used for government projects (e.g., armaments). To this day, it is still anchored in many people’s minds and in standards for projects in regulated environments (e.g., medical technology, banks). This leads to disputes in teams that prefer agile development processes. This article helps to resolve…
Details
Laws and standards formulate requirements on how medical device manufacturers must define and document the development process. Notified bodies check these requirements during audits. This article on the development process provides tips on how to design the process and how to align it with other processes, such as the risk management process.
Details
Medical software manufacturers must meet the legal requirements for software components in order to “approve” their devices. This article presents these requirements and gives seven tips on how to fulfill them quickly and easily.
Details
Since 2020, the German legislature has allowed the reimbursement of digital health applications (DiGA). DiGA manufacturers must fulfill several requirements for this. This article describes the steps required to do so.
Details