IVD medical device validation confirms the device’s medical purpose. IVD medical device verification, on the other hand, proves whether the IVD works as intended.
In this article, we provide a five-step guide on how to carry out the verification and validation of your IVD medical devices in a targeted manner and without unnecessary effort. We also show you the most common mistakes and how to avoid them.
1. Definition and differentiation of terms: validation of IVD medical devices, verification, performance evaluation, qualification, process validation
a) Definitions of terms
With a multitude of terms such as “verification,” “validation,” “performance evaluation,” “analytical performance evaluation,” “clinical performance studies,” etc., it is often difficult for IVD medical device manufacturers to understand the context or the differences between the terms. If you feel the same way, the following section can give you a quick overview.
The terms listed have one thing in common: They are used to demonstrate the safety and performance of an IVD medical device. Furthermore, they apply equally to all types of IVD: IVD devices, IVD assays, reagents and kits, and IVD software.
The term “validation” is defined in ISO 9000:2015 as follows:
“Confirmation, through the provision of objective evidence that the requirements for a specific intended use or application have been fulfilled”
Source: DIN EN ISO 9000:2015
For an IVD medical device, the “specific intended use,” as defined in general terms in ISO 9000:2015, is referred to as the “intended purpose.” Manufacturers use the intended purpose to determine what an IVD may be used for. This includes, for example, the medical purpose, the type of information provided, the sample type, the patient population, the intended users, and the use environment. The validation of IVD medical devices is, therefore, the check of whether the device fulfills the intended purpose, including the attributes mentioned.
ISO 20916:2019, which relates to good study practice in clinical performance studies for IVD medical devices, defines “validation” with direct reference to the intended purpose:
“verification that the specified requirements are adequate for an intended use”
Source: ISO 20916:2019
In contrast, verification always refers to the check of individual product requirements and product design specifications (design input requirements, design specifications) and is defined as:
“confirmation, through the provision of objective evidence, that specified requirements have been fulfilled”
Source: DIN EN ISO 9000:2015
“confirmation by examination and provision of objective evidence that the specified requirements have been fulfilled”
Source: ISO 20916:2019
The term “performance evaluation” originates from the EU Regulation 2017/746 on in vitro diagnostic medical devices (IVDR). According to IVDR, Annex XIII, the performance evaluation of IVDs comprises three elements:
- Scientific validity of an analyte
- Analytical performance
- Clinical performance
Analytical performance refers to the “ability of a device to correctly detect or measure a particular analyte” (see IVDR, Article 2 (40)). The applicable analytical performance parameters are specified by manufacturers as product requirements for the respective IVD (see IVDR, Annex I, Section 9.1.a)). Accordingly, the verification of the analytical performance is equivalent to the verification of the entire device (see below).
On the other hand, the inspection of clinical performance is part of the validation of the IVD medical device. It is defined as the “ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user” and, thus, demonstrates the intended purpose of the IVD.
Manufacturers should note that the performance evaluation of an IVD differs significantly from the clinical evaluation of a medical device.
Read our detailed article on the performance evaluation of IVD medical devices.
This article provides an overview of the clinical evaluation of medical devices.
Do you have IVD medical device software? In this article, we have summarized the special requirements for the clinical evaluation of medical device software and the performance evaluation of IVD software.
b) Differentiation from the qualification of systems and devices (DQ, IQ, OQ, PQ)
The medical device regulations do not differentiate between DQ, IQ, OQ, and PQ. However, it is advisable and necessary for manufacturers to deal with the qualification of devices and systems.
To carry out verification tests and validation studies, manufacturers may need other devices and measuring equipment in addition to the IVD medical device to be checked. They must be able to rely on the functionality and suitability of these devices for the intended purpose (e.g., measurement of a pH value, determination of a nucleic acid concentration, extraction of DNA from blood samples). To check suitability, they carry out a qualification of the required equipment and measuring equipment. The equipment and devices used to produce an IVD medical device must also be qualified for their intended use.
A distinction is made between the following qualification activities:
- Design Qualification – DQ
Was the device developed as specified (in the system architecture), and was the system assembled from these components? - Installation Qualification – IQ
Is the device or system complete, and can it be put into operation as described? - Operation Qualification – OQ
Are all functions available as specified? Does the device or system function under the specified environmental conditions? - Performance Qualification – PQ
Does the device or system fulfill its function with the specified accuracy even at the performance limits and in the event of misuse?
Medical laboratories – the “users” of in-vitro diagnostics – must also carry out a qualification of their “laboratory equipment” before using it for the first time. This is required by both ISO 15189 and the guidelines of the German Medical Association (“Bundesärztekammer”, RiLiBÄK). Laboratory personnel qualify a device to ensure that it is suitable for the intended work steps (e.g., sample processing, quality control) and provides the required performance. ISO 15189 further explains that the term laboratory equipment includes “hardware and software of instruments, measuring systems, and laboratory information systems.” This means that the topic of Computerized Systems Validation (CSV) is also relevant.
ISO 15189:2023-03, Chapter 6.4
c) Differentiation from process validation
During process validation, the aim is to prove that the process always leads to reproducible outputs or devices in accordance with previously specified quality characteristics.
“Process validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.”
Source: FDA 21 CFR 820.3
Regarding Good Manufacturing Practice (GMP), process validation is crucial in producing an IVD medical device. Once manufacturers have established the manufacturing process and documented it based on manufacturing regulations and instructions, they examine the individual production steps for possible risks. Based on this risk analysis, the manufacturer determines which process steps are critical and may need to be safeguarded and validated with quality controls.
The processes for sterilizing an IVD medical device must also be validated.
2. Regulatory requirements
The verification and validation of IVDs are intended to demonstrate the benefits of an IVD. However, what legal requirements must IVD medical device manufacturers adhere to in their verification and validation activities?
Article 5 of the IVDR specifies the requirements that IVD manufacturers must fulfill before placing an IVD on the market or allowing an IVD to be put into service by the user. Thus, manufacturers should first specify the general safety and performance requirements relevant to a device in accordance with Annex I of the IVDR by considering the intended purpose of the IVD. Manufacturers must prove that the device meets the specified safety and performance requirements (see definitions). To do this, they should perform a performance evaluation according to Article 56, i.e., verification and validation.
“Freedom from unacceptable risks”
Source: ISO 20916:2019
“‘performance of a device’ means the ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose;”
Source: IVDR, Article 2 (39)
The following standards describe further criteria for the verification and validation of an IVD medical device:
- ISO 13485:2016 requires in Chapters 7.3.6 and 7.3.7 the performance and documentation of “development verification” and “development validation.”
- For IVD medical device software, IEC 62304:2006 + A1:2015 specifies unit tests, integration tests, and inspection of the software system as verification activities in Chapters 5.5. to 5.7.
- IEC 82304-1:2016 Chapters 6.1 to 6.3 deal with the implementation and documentation of validation of standalone software for medical devices. It is applicable to standalone software for IVD medical devices.
- The aforementioned standard ISO 20916:2019 specifies the requirements for conducting and documenting performance evaluation studies for IVDs and is referenced in the IVDR.
3. 5 steps to IVD medical device verification
Step 1: Define requirements and specifications
The task of verification of an IVD medical device is to check the specified product requirements. Manufacturers must, therefore, have defined these beforehand.
Step 2: Choice of verification activity
Manufacturers can use a variety of different tests, experiments, and studies for verification. The choice of verification activity depends on the type of IVD medical device (e.g., assay, device, software) and the device or software requirement to be checked. Verification can be performed using human patient samples, a synthetic sample matrix, or in silico. For the verification of IVD medical devices, manufacturers can use real patient data for testing and simulated data or computer models.
Manufacturers should document the basic test strategy in the product-specific development plan. Finally, detailed test plans describe the exact procedure for conducting experiments or tests.
Step 3: Bottom-up testing of the individual components
As a rule, manufacturers first check their device’s individual parts/components and test whether they meet the specified requirements. This can be, for example, a reagent of a kit whose actual enzyme activity is measured or unit tests for IVD medical device software.
With the component or unit tests, IVD medical device manufacturers ensure that the individual components of their device function as intended. If the testers notice errors in a product component, the developers can correct them.
Depending on the type of IVD medical device, manufacturers carry out different activities:
IVD medical device assay
In an IVD medical device assay, the tests for component verification include, for example,
- measurements to inspect the concentration of a substance in the reagent,
- analyses to check the length and sequence of a primer,
- testing of adequate primer and probe concentrations in a PCR kit,
- optimal antibody concentrations in an ELISA kit.
IVD medical device
During the verification of IVD medical devices, the following is investigated:
- The function of individual device components and their interaction
- Electromechanical safety in accordance with EN 61010-2-101
- Electromagnetic compatibility in accordance with EN 61326-2-6
- Control software and much more
IVD medical device software
The verification of IVD medical device software – i.e., software testing – includes, among other things:
- Unit tests
- Integration tests
The validation of any tools used for testing is subject to Computerized Systems Validation (CSV).
Read more about the use of artificial intelligence in medical devices, the special requirements for AI-based medical device software, and the validation of machine learning libraries.
Step 4: Verification of the device – system tests
Only when the individual components of a product meet their specified requirements should manufacturers test the entire device – i.e., the final IVD medical device kit, the analyzer, or the standalone software. IVD medical device software is referred to as system testing. The testers check the interaction of the individual product and software components in the final device.
The verification of the entire device includes, among other things, the analytical performance evaluation to confirm the analytical performance of the IVD (see IVDR, Annex I, Section 9.1.a). Manufacturers always verify the finished end product to answer the following questions, for example:
- How consistent are the outputs under constant conditions (repeatability)?
- Do the environmental conditions influence the output (reproducibility)?
- How accurately can extreme values be determined (detection limit, quantification limit)?
- Which sample properties, including pathological changes and drugs in the sample, have what influence on the analytical specificity and sensitivity of the IVD medical device (interferences, cross-reactions)?
- Do the outputs differ depending on the user (reproducibility)?
- How does the reagents’ age or type of storage influence the output (shelf life, shelf life after opening, transport stability)?
- Does the device work safely and without compromising its performance with other products, reagents, devices, or software systems (compatibility)?
Software manufacturers should also carry out sufficient tests to prove that the software meets the IT security requirements.
Step 5: Investigating the compatibility of the IVD medical device with other devices
A basic safety and performance requirement, according to Annex I of the IVDR, states that the combination of an IVD with other devices or systems must not compromise the intended performance (see Annex I, Section 13.1.).
“If the device is intended for use in combination with other devices or equipment, the whole combination, including the connection system, shall be safe and shall not impair the specified performances of the devices. Any restrictions on use applying to such combinations shall be indicated on the label and/or in the instructions for use.”
IVDR, Annex I, Section 13.1.
Combinations can be, e.g.:
- Use of an IVD medical device assay with a laboratory device
- Compatibility of an IVD medical device with the sample preparation method
- Use of raw data generated by different devices by one IVD medical device software
During the verification phase, IVD manufacturers must check and demonstrate the compatibility and interoperability of the IVD with other devices.
Definitions
‘compatibility’ is the ability of a device, including software, when used together with one or more other devices in accordance with its intended purpose, to:
(a) perform without losing or compromising the ability to perform as intended, and/or
(b) integrate and/or operate without the need for modification or adaption of any part of the combined devices, and/or
(c) be used together without conflict/interference or adverse reaction;
Source: IVDR, Article 2 (18)
‘interoperability’ is the ability of two or more devices, including software, from the same manufacturer or from different manufacturers, to:
(a) exchange information and use the information that has been exchanged for the correct execution of a specified function without changing the content of the data, and/or
(b) communicate with each other, and/or
(c) work together as intended;
Source: IVDR, Article 2 (19)
Requirements of ISO 13485:2016
Like the IVDR, ISO 13485:2016 explicitly requires the verification and validation of a device’s interaction with other systems or products in Chapters 7.3.6 and 7.3.7.
4. 5 steps to IVD medical device validation
Step 1: Defining objectives
The validation of an IVD medical device has the task of checking the intended purpose (intended use).
Step 2: Selecting validation activities
The validation of IVD medical devices is usually based on clinical performance studies. During these “clinical investigations,” the intended users (e.g., laboratory personnel, pathologists, human geneticists, depending on the intended purpose) test the IVD in its actual use environment (e.g., in a medical laboratory). The validation of the IVD is based on samples or data (in the case of software) from patients for whom the IVD is intended according to its intended purpose.
Suppose scientific peer-reviewed literature is available for an IVD in which the clinical performance parameters of this device are documented. In that case, manufacturers may also use these publications as a source in addition to clinical performance studies (see IVDR, Annex XIII, Section 1.2.3.).
For CE-marked IVDs that are already on the market, manufacturers may, under certain conditions (e.g., same intended purpose, patient consent, study plan, raw data available), use data from routine diagnostic use to demonstrate the clinical performance of their IVD (see IVDR, Annex XIII, Section 1.2.3.).
Step 3: Determining the questions relevant for validation
When validating IVDs, the following questions, for example, must be answered to demonstrate the clinical performance of a device (see IVDR, Annex I, Section 9.1.b)):
- How well can the IVD identify diseased individuals (diagnostic sensitivity)?
- How specifically can the IVD medical device detect that a disease-associated analyte is not present (diagnostic specificity)?
- How well does the IVD differentiate between true-positive and false-positive outputs in a patient population with a particular disease prevalence (positive predictive value)?
- How well does the IVD distinguish between true-negative and false-negative outputs in a patient population with a particular disease prevalence (negative predictive value)?
- How likely is it that a positive output occurs in a diseased person compared to the probability that the positive output occurs in a healthy person (positive likelihood ratio, see also negative likelihood ratio)?
Step 4: Conducting clinical performance studies for IVD medical devices
To answer the questions raised in step 3, manufacturers carry out the following activities, among others:
- They plan the clinical performance study, perform a case number calculation, and prepare a clinical performance study plan (Clinical Performance Study Protocol (CPSP)).
- They provide the analysis system (e.g., IVD assay, (IVD) device, and IVD evaluation software) under routine conditions in the medical laboratory.
- They recruit a sufficiently large collective of representative, well-characterized patient samples. In the case of validation of standalone software, they collect representative data. The patients must have given their consent to participate in the performance study.
- They compare the outputs they obtain with their new IVD medical device with a reference standard or comparator method. The routine and CE-marked IVD medical device is the benchmark against which the new device must be measured. Manufacturers must justify their choice of reference standard. In doing so, they consider their device and its intended purpose as well as the state of the art in diagnostic practice.
- Where applicable, they have the normal value ranges for different parameters confirmed with a sufficiently large sample collective of healthy individuals and determine the product-specific cut-off value.
- They conduct the performance study under controlled, documented and reproducible conditions and fulfill the requirements of the IVDR according to Annex XIII, Section 2 and ISO 2019:2019.
Step 5: Checking usability
The validation of IVD medical devices must also include usability. Manufacturers must ensure that the specified users in the specified context of use (e.g., a medical laboratory) can actually achieve the specified purpose – the analysis of samples or the evaluation of data – with the IVD medical device.
To this end, manufacturers must test the usability of all safety-relevant use scenarios in the usability test in accordance with IEC 62366 or FDA Human Factors Engineering. In the case of an IVD medical device, these use scenarios typically include sample preparation, loading the device with samples and reagents, creating worklists, processing and evaluating them, and understanding the instructions for use. In the case of IVD medical device software, the focus during usability validation is on the user’s interactions with the user interface.
5. Avoiding typical mistakes in the verification and validation of IVD medical devices
a) Underestimating the effort
Manufacturers underestimate the time required. From the start of development to placing on the market, the verification and validation of IVD medical devices often take 50% of the time. Recruiting suitable samples for a clinical performance study can often take months.
b) Disregarding the overall process
It is essential in both the verification and validation of IVDs that manufacturers always consider the context of the application of their IVDs. This includes the entire process: from sampling, sample preparation, and analysis to evaluation and reporting. This means that manufacturers must always consider all relevant hardware, software, reagents, and samples as well as neighboring systems such as Laboratory Information Systems (LIS) in their experiments and tests for safety and performance verification.
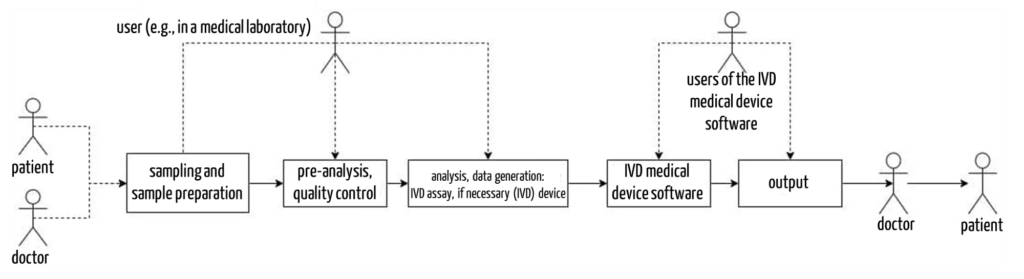
c) Unnecessary V&V activities
The manufacturers must verify the product requirements (Design Input Requirements, DIR). However, many “requirements” are more likely to be classified as Business Requirements (BR), e.g.:
- Reagent consumption
- Waste quantities
- Weight
For approval, manufacturers “only” have to demonstrate implementation of the DIR. A separation of DIR and BR helps to minimize the verification effort.
In addition, manufacturers often do not precisely separate verification and validation and, therefore, carry out inspections twice.
d) Lack of verification and validation activities
Manufacturers do not take usability and compatibility with other hardware, software, and reagents or integration and interoperability with other systems into account during verification and validation of the IVD.
Unfortunately, due to incomplete inspections, inadequate performance, safety gaps, and errors of an IVD may go undetected and defective devices may be placed on the market. As this report in DIE ZEIT (German) shows, this poses enormous risks for patients and hazards to human life.
e) Missing or incomplete specifications and lack of traceability
The specifications and intended purpose should define tolerance limits, which may depend on other parameters. Otherwise, the manufacturer will have to explain any deviations from the specified value.
Manufacturers should not forget to prove that all requirements have been met. This is usually done with the help of a traceability matrix, which links the output of the tests with the requirements that have been proven.
f) Incorrect timing of “official” verification and validation
Manufacturers must verify and validate the “final” devices. Changing the design can render verification and validation results that have already been carried out obsolete. This increases the effort unnecessarily.
Starting V&V activities too late carries the risk of design problems remaining undetected for too long. Early “pre-testing” and an iterative approach during development are therefore helpful.
6. Conclusion
Well-specified product or software requirements provide the basis for the verification and validation activities for an IVD medical device. Manufacturers should ensure traceability between the requirement and the associated verification test or validation study to demonstrate that all product requirements are met.
If manufacturers plan their verification and validation strategy early on, they can identify dependencies between tests and synergies. By avoiding duplication of work and unnecessary repetition, they save time, money, and resources.
By taking a systematic approach to IVD medical device verification and validation, manufacturers ensure they always provide their customers with safe, high-performance, high-quality devices. This saves users a lot of hassle and saves lives.
Reach out to us if you’re looking to demonstrate the safety and performance of your IVD in a targeted manner with minimal effort on your part. We’re here to make the process easier for you.
The IVD team at the Johner Institute will be happy to support you in the planning and standard-compliant documentation of your verification activities and in the validation of your IVD MDs. We can also help you specify the safety and performance requirements for your devices.
If you have any questions about evaluating IVDs’ performance or conducting usability studies, please contact our technical experts – they will be happy to help.
Change history
- 2021-04-21: Article completely revised

