Manufacturers of substance-based medical devices such as seawater nasal sprays, mucous membrane-soothing cough syrups, or osmotically active laxatives face several challenges once the Regulation (EU) 2017/745 (MDR) came into force:
- The new Rule 21 leads to a higher classification of class I substance-based medical devices, which results in the involvement of a notified body.
- The MDR poses higher requirements on clinical data. These also concern the proof of equivalence.
This article illustrates what can be done to overcome the regulatory hurdles and ensure the marketability of your substance-based medical devices beyond the transition periods. This is important to continue providing safe and proven healthcare to patients.
In addition, this article provides you with guidance on what you need to consider regarding new developments or significant product modifications of substance-based medical devices (e.g., changes of formulation or intended use), for which MDR compliance is already required from the deadline of 26 May 2021.
1. Introduction
a) Definition “substance-based medical devices”
Substance-based medical devices are medical devices consisting of substances or a combination of substances. Although they are similar to medicinal products in their presentation and pharmaceutical form, they achieve their main intended effect via a physicochemical and/or physical mechanism of action. Medicinal products, on the other hand, have a pharmacological, immunological, or metabolic mechanism of action.
Source: MEDDEV 2.1/3 rev3
b) Examples for substance-based medical devices
Substance-based medical devices are for example:
- Saline nasal drops or sprays
- Mucous membrane moistening syrups, throat sprays, or lozenges
- Artificial tears
- Vaginal creams or gels to restore the physiological milieu
- Skin creams or ointments for moistening/sealing wounds, scars, or skin diseases
- Oral agents to neutralise gastric acid
- Osmotic laxatives
- Products with a defoaming effect for gas-related gastrointestinal complaints
2. Challenges for manufacturers of substance-based medical devices

a) Obligation to provide evidence based on clinical data
In contrast to many other medical devices, substance-based medical devices usually have clinical functions such as reducing symptoms such as cold, cough, abdominal pain, or constipation, and achieve their effects in direct contact with the human body. Therefore, safety and performance of these devices cannot be proven, or not fully proven, by preclinical or performance data. Rather, this evidence requires clinical data.
While clinical investigations are usually mandatory for class III devices and new developments, existing devices with a low risk profile that have been on the market for a long time were usually not subject to their own clinical investigations under Directive 93/42/EEC (MDD).
Clinical investigations are associated with considerable time and resources required and pose major challenges, especially for smaller and medium-sized companies.
b) Higher classification of substance-based medical devices & involvement of notified bodies
The MDR has introduced Rule 21 as a new classification rule for substance-based medical devices in Annex VIII:
“Devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed by or locally dispersed in the human body are classified as:
MDR, Annex VIII, Rule 21
- class III if they, or their products of metabolism, are systemically absorbed by the human body in order to achieve the intended purpose;
- class III if they achieve their intended purpose in the stomach or lower gastrointestinal tract and they, or their products of metabolism, are systemically absorbed by the human body;
- class IIa if they are applied to the skin or if they are applied in the nasal or oral cavity as far as the pharynx, and achieve their intended purpose on those cavities; and
- class IIb in all other cases.”
This means that all substance-based medical devices which were classified into class I under the MDD will be affected by a higher classification under the MDR. For further marketability beyond the transition periods, the involvement of a notified body will be necessary for the first time.
Read more on the classification of medical devices and the transition periods.
c) Mandatory clinical investigations for certain substance-based medical devices
The key to distinguishing between medicinal products and medical devices lies in the correct interpretation of the mode of action by which the products achieve their principal intended effect. Whereas medicinal products have a pharmacological, immunological, or metabolic mode of action, medical devices are defined by their chemical and/or physical mechanism by which they achieve their principal effect. This is also reflected in their intended purpose.
If substance-based medical devices contain pharmaceutically active substances which support the intended principal intended effect, they are assigned to class III according to Rule 14 of the MDR. For these devices, just as for all other class III substance-based medical devices, clinical investigations are mandatory.
d) Increased requirements for the proof of equivalence
According to the MDR, a clinical evaluation based purely on literature can only be carried out if there is sufficient equivalence to a reference product. The proof of equivalence has already been significantly tightened with the MEDDEV 2/7.1 rev. 4 guideline and now also with the MDR, compared to the specifications of the MDD.
According to Annex XIV Part A of the MDR, the following technical, biological, and clinical characteristics are used to demonstrate equivalence:
Clinical equivalence
The device
- is used for the same clinical condition or purpose (including similar severity and stage of disease),
- is used at the same site in the body,
- in a similar patient population,
- has the same users, and
- provides a similar relevant and decisive performance with regard to the expected clinical effect for a specific intended purpose.
Technical equivalence
The device
- is of similar design,
- is used under similar conditions of use,
- has similar specifications and properties,
- uses similar deployment methods, and
- has similar principles of operation and critical performance requirements.
Biological equivalence
The device
- uses the same materials or substances in contact with the same human tissues or body fluids,
- for a similar kind and duration of contact, and
- similar release characteristics of substances.
The MDR further states that the ingredients must be similar in such a way that there is no clinically meaningful difference in safety and clinical performance. Furthermore, it is required that the assessment of equivalence is based on adequate scientific justification.
Note that the medical device and the equivalent product must be identical for some attributes, while for others they only must be “similar.”
e) Difficulties in accessing data of equivalent products
The MDR requires manufacturers to clearly demonstrate that they have sufficient access to the data of the equivalent product in order to demonstrate equivalence.
“It shall be clearly demonstrated that manufacturers have sufficient levels of access to the data relating to devices with which they are claiming equivalence in order to justify their claims of equivalence.”
MDR Annex XIV
For implantable devices and class III devices the following applies:
“[…] the two manufacturers have a contract in place that explicitly allows the manufacturer of the second device full access to the technical documentation on an ongoing basis, […]”
MDR Article 61(5)
The MDCG 2020-5 guideline states that a corresponding contract is not required for all other devices:
“For devices other than implantable devices and class III devices there is no MDR requirement of a contract between the manufacturers for regulating the access to the technical documentation.”
MDCG 2020-5
The MDR requirements that require the most interpretation are those for “sufficient levels of access” to the data of the equivalent products and “sufficient clinical evidence.” As it is not yet entirely clear how the notified bodies will interpret these terms in detail, the manufacturer must explain and justify what is sufficient or adequate with regard to the respective substance-based medical device.
f) Vague definition of the scope of clinical data
While the demonstration of clinical performance and safety and the clinical benefit of substance-based medical devices must be based on clinical data, the extent to which this can be considered sufficient depends on the characteristics of the device and its intended purpose:
“The manufacturer shall specify and justify the level of clinical evidence necessary to demonstrate conformity with the relevant general safety and performance requirements. That level of clinical evidence shall be appropriate in view of the characteristics of the device and its intended purpose.”
MDR Article 61(1)
The article on clinical data explains what clinical data is and how it can be collected.
g) New requirements regarding the documentation of kinetics of ingredients
Another new requirement imposed by the MDR on substance-based medical devices which are absorbed by the body or distributed locally in the body and contain a drug component is the documentation of the kinetics of these ingredients. This involves the documentation of absorption, distribution, metabolism, and excretion, similar to the requirements for medicinal products for human use.
“Devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body, and that are absorbed by or locally dispersed in the human body shall comply, where applicable and in a manner limited to the aspects not covered by this Regulation, with the relevant requirements laid down in Annex I to Directive 2001/83/EC for the evaluation of absorption, distribution, metabolism, excretion, local tolerance, toxicity, interaction with other devices, medicinal products or other substances and potential for adverse reactions, as required by the applicable conformity assessment procedure under this Regulation.”
MDR Annex I, 12.2
Therefore, ensure that the kinetics of the ingredients are documented in your biocompatibility assessment.
3. Practical tips
In order to continue marketing your existing substance-based medical devices after the end of the transition periods, we recommend the implementation of the following steps:
a) Check the mode of action of the ingredients of your substance-based medical device
Match the mode of action with the formulation and the intended purpose. If there is a physicochemical and/or physical mechanism of all ingredients responsible for the intended purpose, you can make a clear distinction from pharmaceutical effects. This can ensure that your substance-based medical device is not unintentionally classified in class III. For devices in this class, the MDR demands clinical investigations.
While some substances have a clearly determinable mode of action, e.g., in the case of herbal substance mixtures, the type of preparation can play a decisive role in the resulting mechanism, for other ingredients, there is a threshold value for the pharmacological effect which you should consider in your formulation.
Contact us if you need help in determining the mode of action or the differentiation from medicinal products.
b) Review existing (clinical) data
Review existing (clinical) data from the following sources:
- Trials with own product/studies with equivalent products from literature: Check equivalence when using literature data according to the requirements of MDR and the MDCG 2020-5 document on clinical evaluation and equivalence. Document that you have sufficient access to equivalent product data and that you have sufficient data to make an equivalence assessment.
- Post-Market Surveillance (PMS) data: These clinical data can help to demonstrate the safety of your existing substance-based medical device if you have sufficient marketing time and an established quality management system.
- General PMCF activities: The general PMCF activities listed in the PMCF plan and report which are required by the MDR for each product include, for example, pooling clinical experience, obtaining user feedback, and reviewing scientific literature and other sources. The evaluation of these activities provides further clinical data for your device.
c) Identify missing data using a gap analysis
Determine which functions of your substance-based medical device can be demonstrated using clinical data and which can be demonstrated using performance data. In the light of the device’s characteristics and intended use, specify the amount of clinical data you consider sufficient for your existing device.
d) Generate missing preclinical/in vitro data to demonstrate biocompatibility and/or non-clinical performance functions/claims
If preclinical data or in vitro data are suitable to fill existing gaps in demonstrating the safety and performance of your substance-based medical device, you should generate them now.
e) Generate missing clinical data for your existing substance-based medical device via a PMCF study, if required
According the MDCG 2020-6, there is scope for discretion as to whether or in which cases PMCF studies for legacy devices are required:
“In some cases, it may be necessary for the manufacturer to undertake PMCF to generate new data for these legacy devices prior to CE marking under the MDR, whereas in other cases (…) it may be possible to demonstrate conformity with the relevant GSPRs with a more limited clinical data set.”
MDCG 2020-6
If clinical data are needed to fill existing gaps, you should always initiate a PMCF study if
- the existing clinical data are insufficient to demonstrate safety, performance, and clinical benefit, including claims for clinical functions,
- the evidence of performance and/or safety is based solely on preclinical data, or
- there is evidence from clinical/preclinical data of the own device or equivalent products, from risk management, or from the state of the art that safety, performance, and/or clinical benefit require re-evaluation with regard to certain aspects.
4. Conclusion and summary
If you want to continue marketing your legacy substance-based medical devices under the MDR after the end of the transition periods, we advise you to act now in order to be able to meet the regulatory requirements.
Use the time and generate missing (clinical) data. The effort should be based on the risk class and at the same time conserve resources, i.e., be as low as possible and as high as necessary to ensure patient safety. The different possibilities for generating data for substance-based legacy devices depending on the risk class are summarized in Fig. 2.
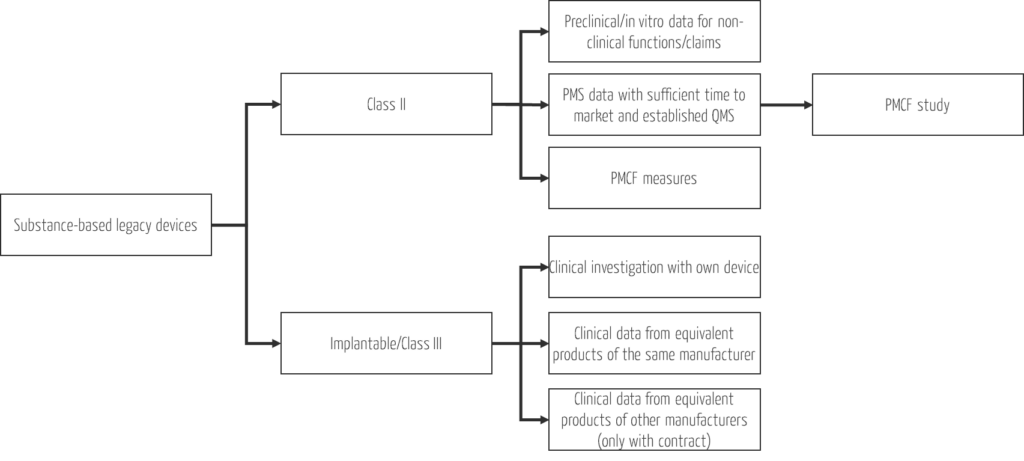
While clinical investigations are mandatory for class III devices and new developments, you have simple, effective, and resource-efficient options for generating clinical data for all other existing substance-based medical devices. Take the opportunity to start PMCF studies for these devices when existing clinical data are insufficient to demonstrate safety and performance of clinical functions.
The Johner Institute supports you in overcoming the regulatory hurdles and ensuring the marketability of your substance-based medical devices.
We answer questions such as:
- Is your substance-based medical device even a medical device? If so, in which class can it be categorized?
- Does your substance-based medical device contain substances that may be considered medicinal products?
- What clinical data do you need for the clinical evaluation of your substance-based medical device? What is the quickest way to collect missing data?
- Does your medical device meet the requirements of the MDR? Can you continue to market your legacy device in compliance with the law?
Contact us or use the Johner Institute’s free micro-consulting service for brief questions. Our experts will also be happy to prepare a clinical evaluation or biocompatibility assessment for your substance-based medical device.
