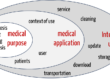
In May 2016, the German version of IEC 60601-1-2:2014 (Edition 4) was published as DIN EN 60601-1-2:2016 with the title “Electromagnetic disturbances – Requirements and tests.”
At the end of 2020, a new version of this “EMC standard” – modified by Amendment 1 and called Edition 4.1 – was published.
Medical device manufacturers who IEC 60601-1-2 applies to should be aware of the changes between the two editions of this standard because they have to decide whether new EMC tests or even a re-design of their devices are required to ensure the conformity of their devices.
The decision diagram in chapter 5 (“Tips”) will help you with this.
1. What is electromagnetic compatibility?
a) Electromagnetic immunity
Medical devices must be safe and effective. For example, they must not fail or malfunction just because a doctor is talking on a cell phone nearby and the cell phone radiation interferes with the medical device. That’s why IEC 60601-1-2 requires “electromagnetic immunity” for medical electrical equipment.
b) Electromagnetic emissions
Conversely, it must also not be the case that “electromagnetic emissions” from medical devices negatively affect their environment, for example, other medical devices, radios, the telephone and wireless networks, computers, or the power supply.
c) Immunity + emissions = electromagnetic compatibility
IEC 60601-1-2 therefore determines the requirements for electromagnetic compatibility (EMC) or, more precisely, electromagnetic disturbance, which is why it is often referred to as the EMC standard.
The standard defines the term electromagnetic compatibility:
“ability of ME equipment or an ME system to function satisfactorily in its electromagnetic environment without introducing intolerable electromagnetic disturbances to other equipment in that environment.”
Source: IEC 60601-1-2
Therefore, the standard is also suitable for meeting the MDR requirement for interoperability and compatibility in relation to the electromagnetic environment and is considered a harmonized standard.
2. Overview of IEC 60601-1-2
a) Scope
Medical electrical equipment
IEC 60601-1-2 represents the state of the art for all manufacturers whose medical devices or accessories fall within the scope of the base standard IEC 60601-1. In other words, medical electrical equipment and systems, ME equipment and ME systems for short.
This means its scope is by definition limited to medical devices and accessories that have an applied part. This is the part of the medical device that must come into physical contact with the patient when the device is used as intended.
However, its applicability is not limited to ME equipment and ME systems. Manufacturers who provide electrical equipment for use in a medical environment, for example, a ward round computer in an intensive care unit or a wall monitor in an operating room that is not intended to be a medical device, should also comply with this standard to ensure that other medical devices are not affected. Often these non-medical devices are marketed under the Low Voltage Directive and are given the additional information “medical grade” in the declaration of conformity.
This article will give you a quick overview of the applicability and requirements of IEC 60601-1.
Implants
IEC 60601-1-2 does not apply to implants (implants have their own standards, e.g., ISO 14117) but does apply to accessories that monitor or control an implant from outside the body.
IVD devices that are also medical devices
The immunity and interference requirements also apply for IVD devices used in a laboratory. However, the applicable standard in this case is not IEC 60601-1-2 but IEC 61326-3-2 for laboratory equipment. Nevertheless, IVD devices that are operated in a medical environment, known as point-of-care IVDs, should use the test severity levels of IEC 60601-1-2.
Both standards, IEC 60601-1-2 and IEC 61326-3-2, refer to the same set of generic standards when specifying the tests: IEC 61000-3 generic standards for emissions and 61000-4 generic standards for immunity. IEC 60601-1-2 and IEC 61326-3-2 give specific limits or test levels for this testing.
Medical devices that also use radio technologies
In addition to the requirements of the MDR, these devices must also meet the requirements of the RED (Radio Equipment Directive). For these devices, following IEC 60601-1-2 alone is not sufficient to meet the requirements of the RED. Therefore, other standards apply, and these are mainly written under the auspices of the European organization ETSI (European Telecommunications Standards Institute) and are available free of charge. It is exciting that these standards take into account not only electromagnetic coexistence but also the biological effects of radiation (biological hazards), which IEC 60601-1-2 completely ignores.
Medical devices operating in special environments
For medical devices operated in ambulances or air ambulances, there are additional requirements, for example, RTCA DO-160 or ISO 14708 for implants in an MRI environment.
b) Requirements of the EMC standard
IEC 60601-1-2 specifies
- which types of electromagnetic interference medical devices must be able to cope with without problems,
- how much electromagnetic radiation medical devices themselves may emit,
- how these requirements may depend on the specified use environment and the risk from the device,
- how manufacturers and/or testing laboratories must plan, test, and document these measurements, and
- how manufacturers should label the device and the information that has to be included in the instructions for use.
c) Structure of IEC 60601-1-2
The structure of the standard is based on these requirements:
- Section 6 specifies how the tests should be documented.
- Section 7 establishes the emissions requirements for electromagnetic radiation.
- Section 8 establishes the immunity (imission) requirements.
- Section 9 specifies the requirements for test reports.
Special attention should be paid to Annex F, which makes clear the standard’s close relationship with risk management (according to ISO 14971). This focus on risk management is new since the 4th edition and is explained even better in Edition 4.1.
3. Differences between the editions of the standard
a) Difference between the 3rd and 4th editions of IEC 60601-1-2
The 4th edition of IEC 60601-1-2 differs significantly from the previous version. This is clear even from a quick look at the table of contents.
The changes include:
- Use of the risk analysis as the reason for establishing immunity test levels
- EMC as an integral part of the risk analysis (effect on basic safety and essential performance characteristics)
- Test levels for immunity depend on the use environment:
- Professional healthcare (practices, clinics, etc.)
- Home healthcare (homes, businesses, public ways and buildings, vehicles)
- Special (MRI, military, heavy industry)
The changes have a significant impact on the design and architecture of medical devices. As with the changes introduced with the 3rd edition of IEC 60601-1, risk analysis has also been given a significant role in the standard in the new version as well.
Manufacturers must accurately determine the essential performance characteristics of their equipment in order to establish reliable performance criteria in the EMC test plan.
b) Difference between Editions 4.0 and 4.1
Amendment 1 published at the end of 2020 amends the 4th version of the standard. Together, the two documents make up Edition 4.1.

Reasons for the changes and an overview
The standards committee identified a total of 15 changes that it felt were so urgent that they couldn’t wait until the fifth edition of the standard:
- New digital technologies (RF transmitters)
The quantity and density of RF transmitters (e.g., WiFi, Bluetooth connections, DECT, RFID, LTE, 5G, wireless charging, etc.) has increased massively. New antenna topologies and higher transmission rates have led to a more complex and diverse use and load pattern. Similarly, the increased portability of equipment has hugely reduced the distance between the sources of radiated RF energy and the equipment that this energy could interfere with.
This has led the authors to increase the immunity requirements for equipment used in close proximity to sources of interference. For example, insulin or patient-controlled analgesia pumps that are worn on the body and whose failure would be unjustifiable. - Comprehensibility of Edition 4.0
Annex F, which was significantly revised in the fourth edition, has obviously raised some questions. As a result, the authorship team has made this annex easier to understand and added comments and examples to each reference to risk management. - Difficulties when applying the standard to large equipment
Large equipment, such as MRI machines, is not easy to test in an EMC laboratory. Therefore, Edition 4.1 provides specific guidance for testing these medical devices. - Obsolete references
IEC 60601-1-2 contains dated references. Since new versions of standards, such as the third edition of ISO 14971, are now available, these references had to be updated.
New test method according to IEC 61000-4-39
For testing immunity near typical RF transmitters, for example cell phones, Bluetooth or WLAN, the test methods described in standard IEC 61000-4-3 –Testing and measurement techniques – Radiated, radio-frequency, electromagnetic field immunity test – were previously essential.
However, the authors of IEC 60601-1-2 made it clear in the explanation for Section 8.10 of the 4th edition that testing according to IEC 61000-4-3 alone was no longer sufficient and started to develop an additional test method. But the test method had still not been validated at the time the 4th edition was published.
This test applies specifically to equipment or systems that are directly exposed (< 15 cm) to sources of interference. This should further reduce the risk presented by interference-sensitive parts, such as coils, signal transformers and Hall-effect sensors. Therefore, IEC 61000-4-39 supplements IEC 61000-4-3 and does not replace it.
During testing, interference fields that are characteristic of the typical use environment are simulated. These include:
- Induction cooking appliances and ovens in domestic environments
- RFID transponders and readers
- For example, surgery for identification of instruments
- For example, surgery to avoid swabs or sponges remaining in the patient
- Position detection devices (e.g., in catheter laboratories)
- Wireless power transmission systems for electric vehicles
These new test methods are introduced in Section 8.11 of AMD 1 (“IMMUNITY to proximity magnetic fields in the frequency range 9 kHz to 13.56 MHz”).
This makes it clear that the authors are gradually adjusting the standard to reality so that it continues to represent the state of the art.
Testing of large systems
So far IEC 60601-1-2 offers three methods for testing the immunity and interference of large, fixed ME equipment/systems. The EMC standard defines large as larger than 2 m × 2 m × 2.5 m (for example, radiotherapy equipment or MRI machines).
For equipment of this size, the standard offers the following options:
- Testing as a complete system at a test center
- Testing as subsystems at a test center
- Testing at the operator’s site (in situ) after installation and before commissioning
The authors acknowledge that testing subsystems or testing the system at the operator’s site is often not appropriate or not possible. It can also be the case that countries will only authorize the importing of equipment/systems for which there is proof of a valid EMC test.
AMD 1 therefore now also permits testing at the manufacturer’s site. Manufacturers usually have the auxiliary equipment required for the control and monitoring of the test specimen.
This makes testing in a representative configuration possible, which also fulfills the requirement for testing in operating mode. Testing at the manufacturer’s premises can therefore be seen as equivalent to in situ testing.
When carrying out testing at the manufacturer’s site, good practice for EMC measurement must be followed. If the applicable EMC generic standards permit on-site, in-situ testing, the requirements in the EMC generic standards take precedence.
In addition, an explanation justifying the testing of ME equipment/systems at the manufacturer’s site should be provided in the test plan and documented in the test report.
Manufacturers can commission EMC test centers provided they have the equipment and personnel required to conduct testing at the manufacturer’s site.
Interaction with risk management
The new Annex F contains a table that extends over six pages. It contains the reasoning behind the considerations for all sections of the standard with risk management requirements and describes the order in which risk management activities should be performed.
This should be welcomed as manufacturers have struggled to implement risk management in the context of EMC. Test centers have also found it difficult to monitor risk-minimizing measures.
For example, you should explain how the applied or assumed risk minimization can reasonably be expected to remain effective over the specified operational lifetime and in all specified use environments.
If, for example, you reduce the ESD test levels because your equipment is intended for operation in an environment with controlled humidity, you would need to discuss whether the equipment could also be used outside of that environment. Components such as filter capacitors also age more quickly when they are exposed to higher temperatures or other physical stresses.
Risk analysis is not always about estimating probabilities. Simple yes/no questions are also part of a risk analysis. For example, whether a hazardous situation or the deterioration or failure of a component is likely enough to require consideration. Similarly, defining the EMC environment is part of a risk analysis. Therefore, not all issues have to be documented in the risk table.
4. Transitional periods in the EU and the USA
a) Europe
Transition from Edition 3 to Edition 4
Harmonization was expected around the end of 2016 and the start of 2017.
The fact that Beuth-Verlag had already published DIN EN 60601-1-2 reinforced this assumption.
But even though the harmonization process has come to a standstill, medical device manufacturers should still switch to the 4th edition when developing devices. Modifying the design later would be very costly.
The notified bodies require this version of the standard with reference to the state of the art. If harmonization occurs, new ME equipment will have to meet the requirements of the fourth edition of IEC 60601-1-2 – probably without an additional transitional period.
For ME equipment already on the market and ME equipment that will continue to be produced unchanged, the notified bodies accept existing EMC tests, but require continual updates to the risk management file, which must also include risks from electromagnetic interference.
Note that other standards in addition to IEC 60601-1-2 have changed (such as IEC 60601-1 version 3.1) or will change.
You can find an overview of planned changes to the IEC 60601 family of standards here.
Transition from Edition 4 to Edition 4.1
The EU recently published draft standardization requests to the European standardization organizations and in them listed, for all MDR and IVDR devices, the standards that the standards committees must harmonize (write Z annexes for) by May 27, 2024. However, it only includes the 4th edition of the EMC standard.

As only a few standards have yet been harmonized under the MDR, manufacturers have to take into account the state of the art, as described above. As AMD 1 is already listed with the FDA with restrictions, it may accurately reflect that state of the art.
The Johner Institute, therefore, recommends using AMD 1, i.e. Edition 4.1 of IEC 60601-1-2. In addition, the standard is also more in line with reality and therefore makes medical devices more reliable, even if some experts claim that it is still out of touch with reality.
The authors are probably aware of this, but also aware that a tightening of the standard must not lead to a shortage in the supply of medical devices.
b) USA (FDA)
Transition from Edition 3 to Edition 4
The FDA has accepted the 4th edition of IEC 60601-1-2 with minor exceptions since 2014. Since December 31, 2018, the application of the 4th edition of the EMC standard has been mandatory.
In the “Design Considerations for Devices Intended for Home Use” guidance document, the use of the 4th edition for devices used in the home is already recommended, at least with regard to the test criteria.
The application of version 3.1 of IEC 60601-1 became mandatory earlier, from August 01, 2016 to be precise.
Transition from Edition 4 to Edition 4.1
The FDA already lists AMD 1 in its list of recognized standards. Other declarations of conformity will no longer be accepted from December 17, 2023.
In June 2022, the FDA published its 20-page guidance document Electromagnetic Compatibility (EMC) of Medical Devices. In it, the FDA requires RFID systems to be tested in accordance with AIM 7351731.
With hundreds of standards, laws, regulations, directives, and guidelines, it is hard to keep track of new and modified requirements.
Use the Johner Institute’s Regulatory Radar that does this continuous research for you and proactively informs you of what you need to know. This not only saves you work and costs, it also helps ensure you meet the legal requirements and avoid regulatory problems.
5. Tips on how to avoid regulatory problems
These tips include general tips that you should always take into account with regard to PMS activities whenever the standard changes, irrespective of the version and tips that relates specifically to the switch to Edition 4.1.
Tip 1: Bring your documentation up to date
To demonstrate an EMC-compliant design process, you need to document your thinking in an EMC concept. The EMC concept should include, for example, an analysis of the environment and the identification of typical sources of interference, including other medical devices, in the specified environment. It should assess the risk resulting from all possible malfunctions or interruptions. You can find an example of an EMC concept in our practical guide to IEC 60601-1 (German). Additional information can be found in the guidance document “Electromagnetic Compatibility for Functional Safety.”
You may want to review or add to the intended purpose, for example, by specifying more precisely the use environment or whether the equipment is portable or worn on the body.
Check the information in the instructions for use against the new specifications of the EMC standard.
Tip 2: Carry out risk management
Carry out a gap analysis of your existing risk management files against the changed and new requirements of the current versions of the standards. The outcome of the risk analysis review should be:
- you don’t have to do anything or
- you only have to repeat the test to demonstrate conformity or
- you have to modify the device and also repeat the test
Document the results in the PMS report and in the PSUR report, if necessary.
Annex F contains, in Figure F.1, a recommendation for the sequence of risk management activities. You could also incorporate the activities directly into your development plan to show that your design process is EMC-compliant.
Tip 3: Agree the procedure with notified bodies
If you want to continue producing existing equipment unchanged, you should discuss the results of the risk analysis and your planned procedure with your notified body. Clarify with the notified body what actions it expects from you if, for example, you make small modifications to a device.
Tip 4: Test electromagnetic compatibility and identify gaps
Carry out preliminary EMC tests with existing ME equipment and determine whether the current design still meets the modified requirements. The laboratory where you perform this preliminary test does not necessarily have to be accredited.
Tip 5: Check which devices are affected
Manufacturers must take Table 11 of Edition 4.1 into account when planning testing for ME equipment and systems and their accessories.
No testing is required if the equipment:
- does not have any components or circuits that are sensitive to interference or
- whose disturbance (according to Table 11) does not result in a loss of basic safety or the essential performance characteristics or
- is guaranteed due to its design to be a minimum of 15 cm from a source of interference
However, if testing is not required, you must justify and document this in the EMC test plan.
Even if the new test in Section 8.11 does not have to be performed for your equipment (see prerequisites), we still recommend conducting the test. This will help you get to know your device better (particularly if the documentation is thin or the original developers are no longer around), and you can also explore the “design reserves,” for example, by raising the test levels even higher than those specified in Table 11. The results will also provide you with additional decision criteria for your product road map.
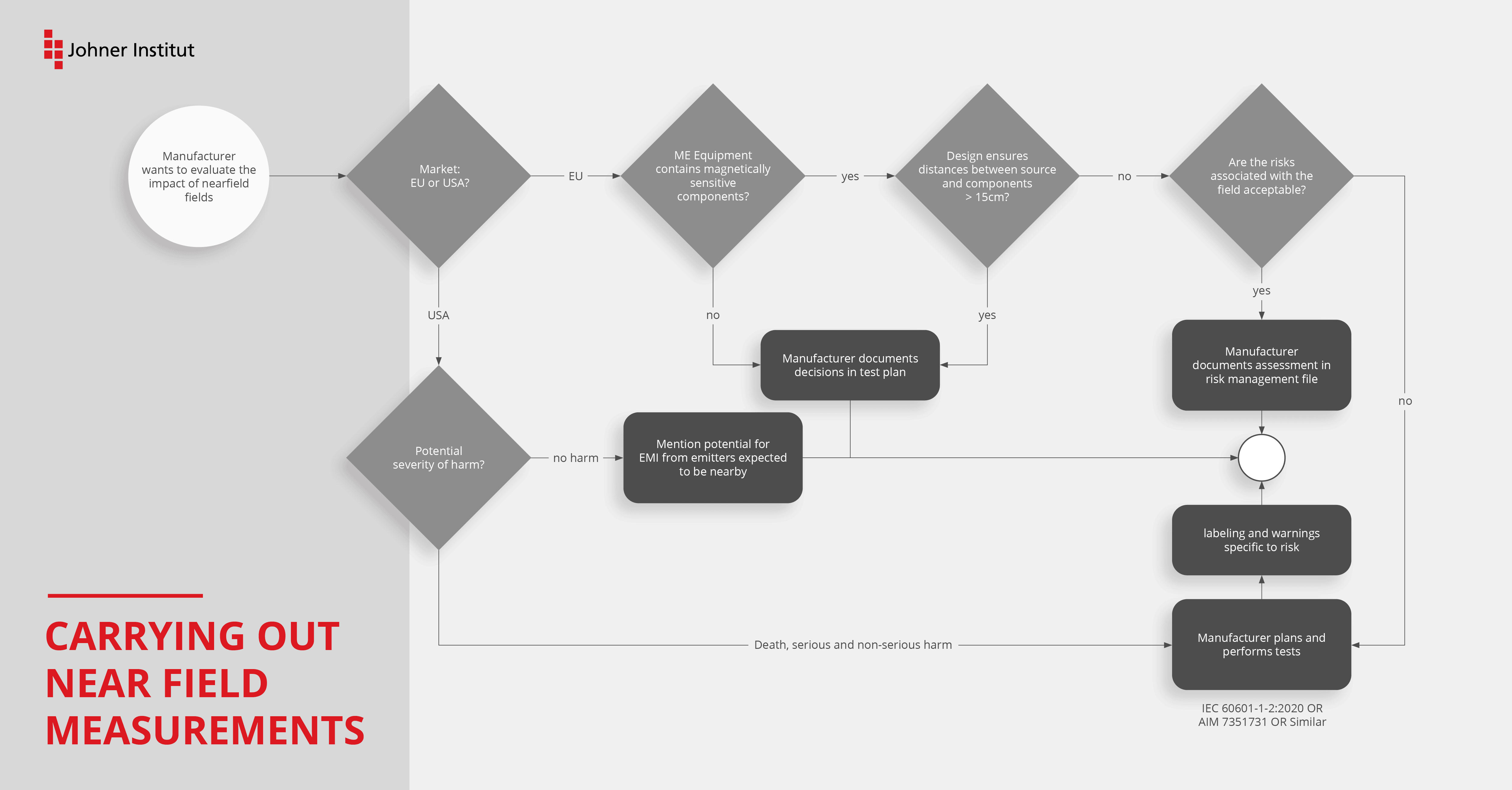
Tip 6: Specify and implement risk mitigations
Create an EMC test plan that complies with the specifications of EN 60601-1-2 (4th edition).
You should also perform a gap analysis to make sure that you are complying with the modified requirements as well. This applies in particular to the requirements for device design and technical documentation.
Depending on the results, check your device and your documentation for completeness and accuracy according to the ER checklist in the MDD and GSPR checklist in the MDR. Use Table ZZ.1 in Annex ZZ of the EN version of the standard.
Derive a list of actions for the review of the architecture (incorporating the risk analysis) from the results of items 2., 3., 7., and 8.
Implement the measures and have your device tested in an accredited testing laboratory.
Declare conformity.
The Johner Institute helps manufacturers check the conformity of their medical devices and their documentation and to restore this conformity with lean and precise actions if there are any deviations. To do this, we create gap analyses, check your documents, create test plans, and help create arguments you can use with authorities and notified bodies.
Contact us (e.g., via the contact form), so that we can work together to define a specific procedure and ensure the conformity and safety of your devices.
6. Conclusion, summary
Amendment 1 improves the previous fourth edition of IEC 60601-1-2 and, together with it, forms Edition 4.1.
This version of the EMC standard is a logical development that was necessary to take technical trends into account and ensure that the standard is comprehensible and up-to-date.
For manufacturers, this new edition makes clear that the state of the art is constantly evolving. They must take this into account, and should always take a risk-based approach to avoid both unnecessary costs and regulatory trouble.
Ultimately, the goal is to ensure the safety and performance of medical devices and thus ensure the safety and health of patients.
Change history
- 2023-03-03: Decision diagram added
- 2021-03: Article from 2016 completely redesigned