Post-Market Surveillance (PMS) is a proactive and systematic process to derive necessary corrective and preventive actions (CAPA) from information about medical devices already placed on the market.
We have created a Post-Market Surveillance checklist for you. With this checklist, you can find out whether you meet the regulatory requirements of the MDR for Post-Market Surveillance of your devices. You will get an overview of all the tasks you need to do to be MDR compliant. This will increase your safety to be successful in the audit and approval of your devices.
Click here for the checklist.
Please also note the other articles on the post-market phase.
1. Definition of Post-Market Surveillance
Both the Medical Device Regulation (MDR) and the In Vitro Diagnostic Medical Device Regulation (IVDR), as well as the FDA, define the term “Post-Market Surveillance.”
“all activities carried out by the manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from their devices placed on the market, made available or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions“
The FDA’s definition is comparable:
“The active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device. “
Source: FDA 21 CFR part 822
The FDA’s definition may be conveniently short. However, the MDR’s more long-winded explanation seems to be more helpful as it not only describes actions but also objectives of post-market activities.
2. Post-Market Surveillance: Objectives
Before bringing medical devices to market, manufacturers must minimize their risks and ensure patients’ safety. This is monitored by authorities and notified bodies during authorization and conformity assessment procedure.
However, some risks manifest over time, when medical devices are used daily by practitioners.
Post-Market Surveillance aims at
- systematically identifying these risks when occurring during practical usage,
- monitoring the devices’ performance afield,
- detecting device faults and safety issues left undetected,
- constantly updating the benefit-risk assessment, and
- quickly initiating necessary measures such as recalls.
Only due to a constant systematic market surveillance (Post-Market Surveillance) can manufacturers guarantee that medical devices provide the promised benefit to patients as well as the lack of any unrestrained risks.
Please read below which data you can considered for market surveillance.
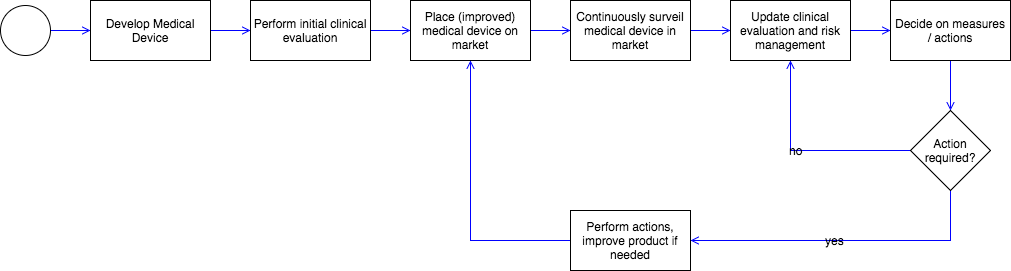
3. Distinctions
a) Post-Market Clinical Follow-up (PMCF)
Post-Market Surveillance aims to continuously verify the benefits of medical devices and to identify previously unknown risks by observing and analyzing daily practical usage.
Closely related to monitoring after placing on the market (PMS) are the terms “Post-Market Clinical Follow-up” (PMCF) or – in the case of IVDs – “Post-Market Performance Follow-up” (PMPF) and vigilance.
“A continuous process to update the clinical evaluation referred to in Article 61 and Part A of this Annex“
Source: MDR
“Continuous process by which data are assessed and analysed to demonstrate the scientific validity, analytical performance and clinical performance of that device for its intended purpose as stated by the manufacturer“
Source: IVDR
Thus, Post-Market Clinical Follow-up (PMCF) or Post-Market Performance Follow-up (PMPF) is about systematically collecting clinical data. Post-Market Performance Follow-up (PMPF) proactively collects safety and performance and scientific data.
Both PMCF and PMPF, in the case of IVDs, have the objective of answering important unanswered questions about the safety or performance of the medical device. Post-Market Surveillance involves collecting all kinds of significant information from the field, for example, service reports, hotline calls, customer complaints, etc.
The PMCF has the objective of updating the clinical evaluation; the PMPF is to renew the performance evaluation of IVDs. Post-Market Surveillance aims to decide on necessary actions to ensure patient and user safety. This decision incorporates the output of the clinical evaluation or performance evaluation. The PMCF or PMPF is thus a subset of Post-Market Surveillance.
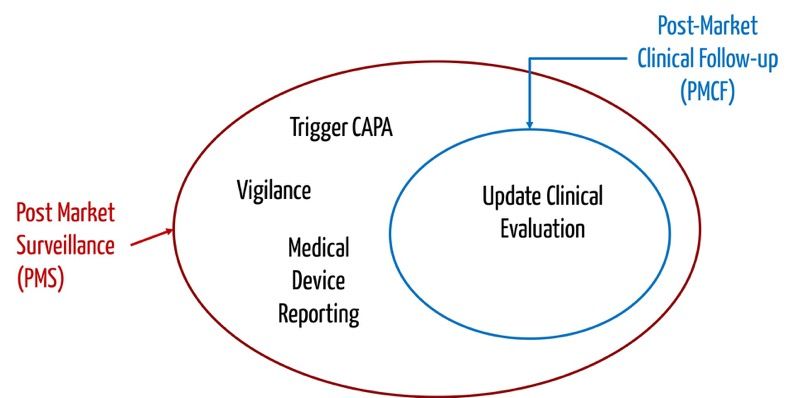
This approach is also reflected in the requirements for the PMS plan in the MDR and IVDR, respectively. They write, for example:
The manufacturer shall undertake to institute and keep up to date a post-market surveillance plan, including a PMCF respectively a PMPF plan.
In contrast, the FDA does not distinguish between the two aspects precisely: it requires, for example, that the PMS plan contain information that is specified for clinical investigations, such as the number of test subjects, the study objective, and consent forms.
Here, you will find tips for writing a PMS plan and on the requirements of ISO 20416.
b) Vigilance
A vigilance system refers to a reactive reporting system: within the vigilance framework, manufacturers must regulate how they report incidents to the competent authorities. Laws and regulations such as the MPAMIV give manufacturers not much freedom in defining the reporting system. As part of proactive Post-Market Surveillance, as a manufacturer, you should also continuously evaluate the data from your vigilance system.
Read more about vigilance here. The article also discusses the distinction between vigilance and post-market surveillance.
c) Market surveillance
Not to be confused with “Post-Market Surveillance” is “market surveillance.” The latter is the task of the authorities.
d) Conclusion
The activities within the scope of Post-Market Surveillance, market surveillance, reporting, Post-Market Clinical Follow-up or Post-Market Performance Follow-ups, and vigilance are partly overlapping. Thus, the terms are often used as synonyms – although they aren’t.
Regulatory requirements usually refer to several aspects.
4. Regulatory requirements
a) Medical Device Regulation (MDR)
The Medical Device Regulation MDR describes the requirements for Post-Market Surveillance in much more detail and concretely than the previously valid Medical Device Directive (MDD, 93/42/EEC). No less than four articles and an annex are dedicated to this topic.
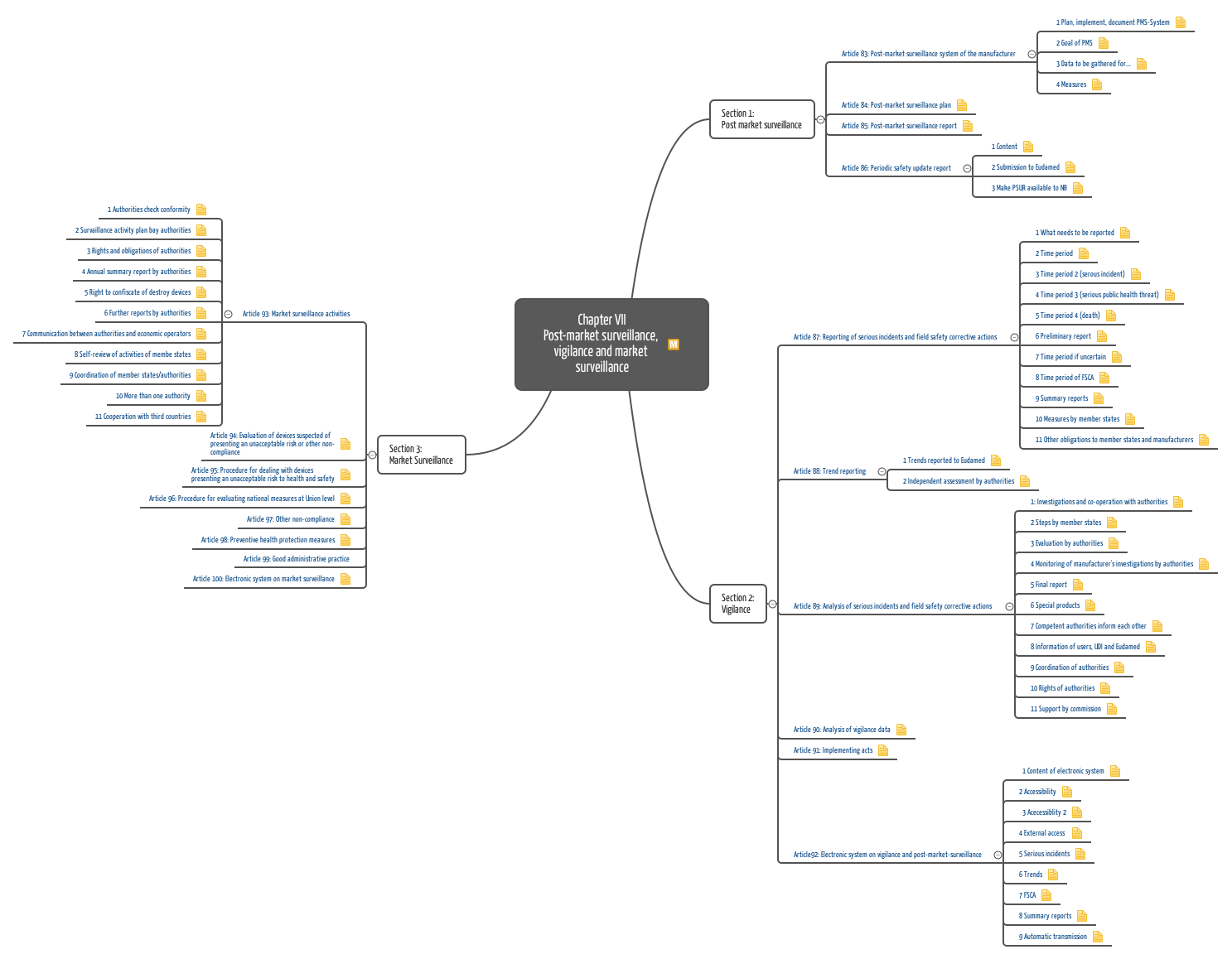
Important requirements of the MDR regarding Post-Market Surveillance are:
- defining, planning, and maintaining the PMS process
- continuously and systematically collecting and evaluating data
- based on these data, continuously decide on measures such as
- initiating CAPA (possibly referring to the device as well as the manufacturer and his processes)
- informing authorities and users
- initiating recalls
- updating the clinical evaluation file and/or risk management files (especially as an output from PMCF activities)
- reporting on results (PMS report or Periodic Safety Update Report PSUR)
Annex III contains the requirements for the PMS plan and, thus, which sources to analyze, how to evaluate them, and how to take action. Specific requirements for the PMCF plan and activities are presented in Annex XIV, Part B.
The article on trend analysis in post-market surveillance provides tips for overcoming common difficulties.
The requirement in Article 83(4) for the manufacturer to inform authorities or notified bodies of derived measures only applies to safety-relevant CAPA, as specified in MPDG §85.
b) Regulation for IVD (IVDR)
The IVDR also describes detailed requirements for the PMS system in four articles (Articles 78-81) and in Annex III. These are almost identical to the above-mentioned requirements of the MDR but differ with regard to PMPF to update the performance evaluation. In addition to continuously ensuring the benefit-risk balance, the focus is also on providing clinical evidence.
Manufacturers are required to assess the current state of the art, confirm scientific validity, e.g., by screening the latest scientific literature, and demonstrate continuity of analytical and clinical performance of their IVD, e.g., through participation in interlaboratory studies, epidemiological studies, database searches, or PMPF studies.
Part B of Annex XIII describes the objectives of PMPF and the requirements for planning, evaluation, and documentation.
c) ISO 13485
ISO 13485 obliges manufacturers to guarantee the effectiveness of QM systems a medical devices’ safety amongst other through systematic market surveillance (Post-Market Surveillance).
d) ISO 14971
The norm on risk management too lays down requirements about the “subsequent phase”. However, the norm’s focus is not reporting. Rather, the norm is about using information coming from production or following stages (e.g., devices’ usage) to assess
- if probability and severity of possible damages are accurately judged
- if risks are fully identified
- if presumed risk acceptance criteria and risk-benefit ratios are valid
Manufacturers acting under ISO 14971 must not only search for problems in the subsequent phase. All information aiding to verify or falsify the own assumptions’ correctness is significant.
Please read below which data you can consider for market surveillance.
The risk management report must confirm that the planned activities are appropriate.
e) USA/FDA: 21 CFR part 822 and “Post-Market Surveillance” guidance document
In 21 CFR part 822, the FDA provides precise regulations on:
- when Post-Market Surveillance is necessary
- what manufacturers need to consider in their PMS plan
- which documents the manufacturers have to produce
- how quickly manufacturers must react in the event of a problem or the FDA must act
The FDA has published a guidance document in 2022, specifically on 21 CFR part 822, to provide further guidance to manufacturers.
Please refer to this article on 21 CFR part 822 (German) and the aforementioned Guidance Document for more information on FDA requirements.
f) ISO TR 20416
The “Technical Report” ISO TR 20416 describes in even more detail how manufacturers can plan and implement PMS. However, it is important to note that a PMS plan, according to ISO TR 20416, does not include all elements required by the MDR or IVDR.
Check out this article on ISO 20416 for an overview of the standard and a suggested chapter structure for PMS plans.
g) Conclusion
In contrast to the reporting system (vigilance), the European specifications for the actual “monitoring after placing on the market” have not been so action-guiding. This has now changed with the new MDR and IVDR, which describe Post-Market Surveillance in a much more comprehensive, concrete, and detailed manner. The following detailed tips will help you with the implementation.
The Johner Institute’s Post-Market Radar can support you in Post-Market Surveillance.
5. SOP and checklist
As a manufacturer of medical devices, you should structure your Post-Market Surveillance, e.g., through respective standard operation procedure SOP, subject to the following aspects:
- Activity Triggers
In general, there are two possible triggers- based on a point in time or period (e.g., once every quarter, every third Monday of a month)
- occasion-related triggers (e.g., customer calls, a new norm was published, 10,000th device was shipped)
- Sources of Information
Examples for sources of information are- customer feedback including customer complaints
- service reports
- assessment results
- observations of employees
- data bases of authorities with reports on problems or measures by manufacturers of comparable devices, technologies or techniques
- calls to the hotline
- specialized clinical literature
- results of PMPF studies
- fairs and conferences
- Activities
Activities typically involve- collecting data
- analyzing data
- evaluating data
- taking actions (e.g., recalls, reporting to authorities, CAPA) or justifying inactivity)
- Responsibilities
Process instructions should also describe which activities are assigned to which roles. For example, medical device advisors, safety officers, risk managers, developers, support specialists, hotline services, and management are involved. - Documentation and Tools
As manufacturers evaluate comprehensive data when conducting Post-Market Surveillance, we recommend documenting this information using tools. Do not forget to validate these tools, which is required under ISO 13485.
Download our checklist as a PDF document here. It details all the tasks you must complete to establish an MDR-compliant PMS process. It will help you pass the audit and increase the safety of your devices.
Ensure a stress-free audit experience: the Medical Device University provides not only video training on Post-Market Surveillance but also proven templates to help you set up your PMS system quickly and cost-effectively.
Change history:
- 2022-12-06: FDA guidance addition to 21 CFR part 822 and general update.


