We have revised this article for you and taken into account the Regulation – EU – 2024/1860 amending IVDR published on the June 13, 2024.
In December 2021, the EU extended the transitional periods of Regulation 2017/746 on in vitro diagnostic medical devices (IVDR) (Regulation 2022/112). In March 2023, the European Commission decided to abolish the “sell-off period” for IVDs that comply with Directive 98/79/EC (IVDD) (Regulation 2023/607).
In January 2024, the European Commission published a proposal to amend the IVDR, which further extends the transition periods. The main reason given for this is the lack of notified bodies.
Although this does not change the date of application of the IVDR, which remains May 26, 2022, the extended deadlines give manufacturers and notified bodies more time to get IVD products through the IVDR conformity assessment and safe and efficient products are not unnecessarily disposed of.
Continue reading to discover the transition periods in place until 2030 and what implications they hold for you.
1. Overview of the transition periods
a) Purpose of the IVDR Amending Regulation?
In particular, the changes give more time to manufacturers of devices that require a notified body under the IVDR. Such products may still be manufactured and placed on the market after the date of validity of the IVDR on May 26, 2022. However, certain requirements of the IVDR must be addressed immediately. Further details can be found below.
b) Which products are affected by the changes?
First of all, it is important that the new regulations concerning transition periods only apply to existing products already on the market. These are devices that have been declared compliant before May 26, 2022, AND fall into classes D, C, B, or A (sterile) under the IVDR. This means that all products will benefit from an extended transition period, except
- class A products without sterile labeling, and
- new products for which a declaration of conformity was not issued before May 26, 2022.
c) How long are the transition periods?
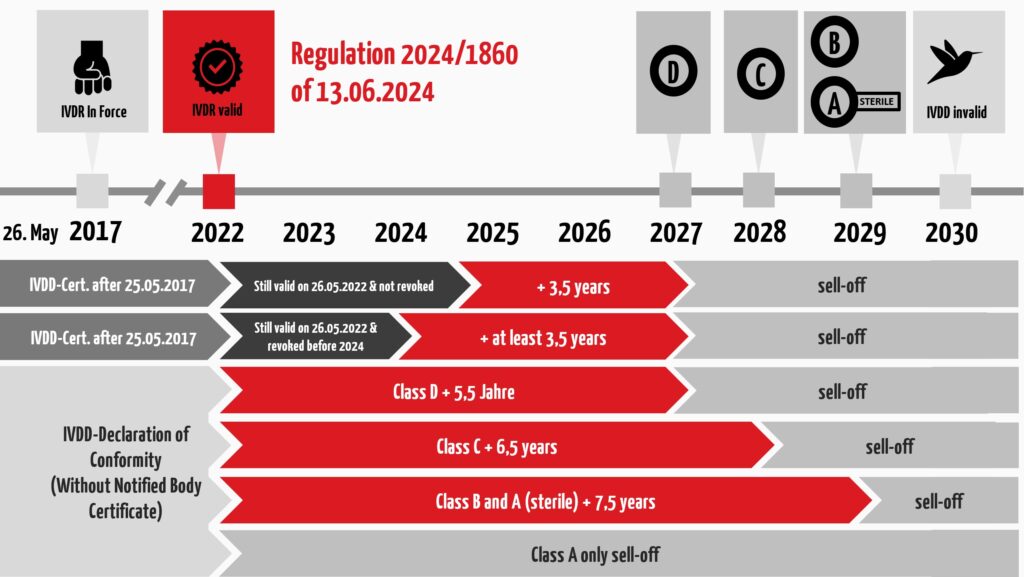
The length of the new deadlines depends on the future IVDR risk class and whether a notified body has already been involved under IVDD. Interestingly, according to the current proposal, the EU has moved the original cut-off date from May 26 to the turn of the year:
- There is no extended transition period for class A (exception: sterile products. These will be given a transitional period for placing on the market until December 31, 2029).
- For class B devices, the same transition period applies until December 31, 2029.
- For class C devices, this deadline ends on December 31, 2028, and
- for class D devices on December 31, 2027.
- The so-called “sell-off regulation” (provision and commissioning) is no longer applicable.
The following figure summarizes the regulations regarding transitional periods under the IVDR. However, please note that these regulations are subject to certain conditions. The subsequent section will provide a detailed description of these.
2. The new deadlines in detail
a) IVD medical devices for which a certificate has been issued by a notified body under IVDD and require a notified body under IVDR
For in vitro diagnostic medical devices for which a certificate has been issued by a notified body under IVDD, the deadline is December 31, 2027 at the latest. These IVDs include devices that comply with Annex II, List A and List B or devices for self-use.
The underlying conformity assessment procedure is of secondary importance. This means that it is not decisive whether the product in question has obtained a certificate in accordance with IVDD through a complete quality assurance system, through type examination or through design testing for self-testing devices.
Let us assume, for example, that you have declared conformity with the involvement of a notified body in accordance with Annex IV of the IVDD (complete quality assurance system) for a List B IVD. The certificate was issued between May 25, 2017, and May 25, 2022. In this case, the certificate validity expires on December 31, 2027. The renewal of the certificate is directly applicable, so notified bodies must not change the date on the individual certificates.
A further condition is that a formal application must be submitted to the notified body by May 26, 2025, and a contract with the notified body must be concluded by September 26, 2025 for the certificate of the IVDD product to remain valid.
However, if the certificate has already expired before the new proposed amendment is adopted, the renewal of the certificate shall be subject to the condition that either the manufacturer has signed a contract with a notified body, or the national competent authority has granted an exemption from the applicable conformity assessment procedure in accordance with Article 54 or has requested the manufacturer to complete the conformity assessment procedure referred to in Article 92 within a specified time period.
If you have voluntarily certified your QM system according to ISO 13485, this certification is not issued by a notified body but by a certification body. A valid certificate, in the context of the IVD Directive 98/79/EC, needs to be issued by an accredited notified body.
b) “Other IVDs” that have already been placed on the market under IVDD without the involvement of a notified body and require a notified body under IVDR
The following new transitional periods apply to in vitro diagnostic medical devices for which manufacturers themselves have issued the declaration of conformity according to the IVDD before May 26, 2022 and which are required to undergo a conformity assessment procedure with a notified body under IVDR in accordance with IVDR classification rules:
- December 31, 2027 for class D IVDs,
- December 31, 2028 for class C IVDs,
- December 31, 2029 for class B IVDs, and
- December 31, 2029 for class A IVDs placed on the market in sterile form.
Depending on the risk class of the IVD, these existing devices may therefore continue to be placed on the market even after the validity date of the IVDR (May 26, 2022), provided that the devices comply with the requirements of the IVDD and there is no significant change in design and intended purpose.
If you want to use this option, please pay attention to the details in the declaration of conformity. This article may help you: EU declaration of conformity.
Example: Assuming that a declaration of conformity in accordance with Annex III was issued under the IVDD without a notified body before May 26, 2022. Then the question arises as to which class the product will fall under the IVDR in the future.
I) If the product falls into class D in the future, you may still manufacture and place these products on the market until December 31, 2027 but you must not significantly change them.
II) If the product falls under class C in the future, its manufacture and placing on the market shall be permitted until December 31, 2028. Significant changes are not permitted.
III) If the device falls under class B or A (sterile) in the future, manufacturing and placing on the market will be permitted until December 31, 2029, which is therefore the longest transition period. Changes to the product are not permitted either.
These transitional periods are also subject to conditions! Manufacturers must have submitted a formal application to the notified body. The deadlines for this are as follows:
I) Class D devices: application submitted by May 26, 2025.
II) Class C devices: application submitted by May 26, 2026.
III) Class B or A (sterile): application submitted by May 26, 2027.
These deadlines are immediately followed by the next condition: 4 months after the above-mentioned application deadlines, a written contract must be concluded with the notified body for the IVDD products to continue to be placed on the market.
The MDCG Guideline MDCG 2022-6 provides you with clarity on the concept of “substantial changes in interpretation and purpose” according to Article 110(3) of the IVDR. No “significant changes” include, in particular, administrative changes and organizational changes – closely aligned with MDCG 2020-03. In addition, the “substantial changes in interpretation and purpose” are defined, with reference to six flowcharts and examples from Chapter 4.3.2.1 onwards.
Please also refer to the article on design changes.
Regardless of the risk classification according to IVDR, you or the distributor can continue to provide and put the product into service even after the end of the transition period, as Regulation 2023/607 has now eliminated the “sell-off rule”.
c) What requirements of the IVDR do manufacturers have to meet, despite the transition period?
However, manufacturers should note that even for devices to which the proposed transition periods would apply, the requirements of the IVDR for post-market surveillance (PMS), market surveillance, vigilance, and registration of economic operators and devices in EUDAMED must be complied with. The proposal of January 2024, which also regulates the transition periods of the IVDR, also obliges economic operators to use each of the 6 EUDAMED modules no later than six months after their publication.
Furthermore, it must now be noted that manufacturers, regardless of whether it is an IVDD device with or without the involvement of a notified body, must implement a quality management system in accordance with Article 10(8) of the IVDR by May 26, 2025.
d) No renewal for (non-sterile) class A devices and “new” IVDs
However, the extensions proposed by the Commission will NOT apply to two product groups:
- (non-sterile) class A devices and
- “new” IVD.
In any case, since May 26, 2022, if the device falls under class A and is not sterile, manufacturers will have toprovide proof of conformity in accordance with the IVDR and comply with all applicable requirements of the IVDR. This means that you will not be able to take advantage of any extended transition period. Since there is no need for a conformity assessment by a notified body, it is solely up to the manufacturer’s resources.
Nor do the extended deadlines apply to “new” in vitro diagnostic medical devices. As such, the EU Commission considers IVDs whose conformity has not been declared under the IVDD.
Since May 26, 2022, manufacturers must also provide proof of conformity in accordance with IVDR for these devices.
e) Inhouse IVD (also called Laboratory Developed Tests, LDT)
Products that are produced and used by medical institutions themselves also benefit from extended transition periods.
However the general safety and performance requirements according to Annex I, IVDR including the performance assessment (IVDR, Article 5 (5) sentence 1, sentence 2 letter a) and sentences 3 to 5) must be fulfilled in the EU since May 26, 2022.
An extension of the deadline until May 26, 2024 is granted for the implementation of the requirements referred to in Article 5 (5), subpoints (b), (c), (e), (f), (g), (h) and (i).
This includes:
- Quality management system
5 (b) refers to the requirement for an appropriate quality management system, which would at least consider the development and manufacturing of IVDs from ISO 13485 in order to comply with the state of the art. - Laboratory quality assurance
5 (c) requires compliance with the requirements of ISO 15189 for quality assurance in the laboratory and the fulfilment of national accreditation regulations, such as the German national RiliBäk (Guideline of the German Medical Association for Quality Assurance in Laboratory Medicine). - Provision of information
5 (e) Providing information to authorities on production, modification, and use. - Public statement of conformity
5 (f) demands the declaration of compliance with Annex I of the IVDR. - Documentation
5 (g) includes the documentation on production, design, and performance, i.e. the “product file”. - Batch record (proof of production)
5 (h) refers to the proof of production according to the specifications in the “product file”. - Serious incidents and corrective actions
5 (i) deals with the gathering and evaluating of experience in clinical application, as well as deriving corrective actions.
For the fulfilment of Article 5 (5, d), the new proposal of the EU Commission grants a transitional period until December 31, 2030. It states:
“In its documentation, the health institution provides a justification for the fact that the specific needs of the target patient group cannot be satisfied or cannot be met at the indicated level of performance by a similar product on the market;“
This requirement was intended to ensure that there is no competition on the market between inhouse IVDs and commercial products, as they are regulated differently. Now, the EU has placed this requirement at the very end of the transition period, reasoning that laboratories can only compare their products with those on the market when all IVD products have been approved under IVDR and registered in EUDAMED.
For further details, please refer to “How the EU regulates medical laboratories”.
f) EUDAMED
Originally, EUDAMED was only intended to be mandatory for all economic operators to be uses after its full functionality. With the EU’s proposal of January 2024, each of the 6 modules is to be used by stakeholders no later than six months after the announcement that the modules comply with the functional specifications. The mandatory use of the first modules relating to economic operators, UDI, notified bodies, and certificates could therefore start in the fourth quarter of 2025. The full functionality of EUDAMED cannot be expected before the fourth quarter of 2027, according to the January 2024 proposal. The final transition period for the use of EUDAMED is the second quarter of 2029.
More information can be found in the article on the EUDAMED.
3. What doesn’t change
The IVDR has been in force since May 26, 2022. This means that the monitoring obligations of products on the market under Articles 78 to 81, as well as on vigilance (Articles 82 to 87) and monitoring by competent authorities also apply, notwithstanding the transitional periods. Thus, the entirety of Chapter VII of the IVDR is to be applied, even if the devices are still being placed on the market under the IVDD.
In addition, economic operators must register within the EUDAMED database.
However, the requirements of this Regulation relating to post-market surveillance, market surveillance, vigilance, registration of economic operators and of devices shall apply to devices referred to […] instead of the corresponding requirements in Directive 98/79/EC.
Proposal of the EU Commission of 14 Oct. 2021 on the adaptation of IVDR (EU) 2017/746
The notified body that issued certificates under the IVDD will continue to be responsible for monitoring conformity during the transition period.
It is also important that no significant changes are made to the products during the transition period, otherwise manufacturers will have to immediately comply with all the requirements of the IVDR.
4. What the changes mean for manufacturers
It is imperative that manufacturers continue to adhere to their implementation plan to ensure that their IVDs become IVDR compliant in time. Precisely because of the scarce resources, manufacturers should not let time pass unnecessarily. Manufacturers should be aware of their IVD’s risk class. Not only the conformity assessment procedure depends on this, but also the transitional period. An IVDR-compliant quality management system must be established by May 2025.
5. Conclusion
The IVDR has been in force since May 26, 2022, and the amending ordinances do not change this. However, the majority of IVD manufacturers are now given more time to provide the necessary proof of conformity for their products. In view of the few notified bodies that currently exist for the IVDR, this should bring great relief for many – and prevent important IVD products from disappearing from the market. In addition, unnecessary disposal of safe products is prevented. For medical laboratories as well, certain requirements can be implemented with a delay. Nevertheless, they must also meet the general safety and performance requirements of Annex I for inhouse IVDs.
Our strategic experts at the Johner Institute will be happy to assist you with any questions you may have about the approval strategy for IVDs or specifically about in-house IVDs. Simply contact us via the form or send us an e-mail.
Change history:
- 2024-10-08: Notes and new transition periods added in accordance with the amending Regulation 2024/1860 of June 13, 2024
- 2024-02-05: Notes and new transition periods from Regulation (EU) 2024/0021 added
- 2023-09-01: Corrections, adjustments to links and editorial changes throughout the article
- 2023-04-11: Regulation Guidance (EU) 2023/607 added
- 2022-03-22: Under 2e) notes on the registration deadline inserted in EUDAMED
- 2022-05-06: Notes on MDCG 2022-6 added
- 2022-01-09: Added note to the Commission proposal


