In vitro diagnostic medical devices (IVDs) are medical devices used to analyze samples derived from the human body. Typically, these products are reagents, kits, instruments, and devices. Software can also be considered as an IVD regarding its intended purpose.
When “approving IVDs,” manufacturers must comply with many regulations, laws, and standards.
The keyword article on IVDs contains the EU and FDA definitions of which devices count as in vitro diagnostic medical devices and must be approved as such. The article also provides an overview of the regulatory requirements for IVDs.
Another article describes the 7 steps to a medical device/IVD. We recommend to read this article first.
1. Significant steps for the approval of IVDs in the EU
The EU Directive 98/79/EC on in vitro diagnostic medical devices (IVDD) was replaced by the “Regulation (EU) 2017/746 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU” – IVDR for short.
Read more about the EU regulation in the article on the IVDR.
Our consolidated version of the IVDR is quite helpful for navigation of the articles and annexes.
If you have placed your device on the market under the In Vitro Diagnostics Directive (IVDD), the IVDR transitional periods are relevant for you.
a) Formulating a precise intended purpose and qualifying the device as an IVD
The intended purpose of a device essentially determines its qualification as a medical device or IVD. It is also decisive for the subsequent classification and possible regulatory approval strategy. As a first step, manufacturers should precisely formulate the intended purpose of their device and comply with the requirements of the IVDR in accordance with Annex I, Section 20.4.1 c) and Annex II, Section 1.1 c). Medical laboratories should also formulate an IVDR-compliant intended purpose for their in-house IVD.
A device must have a medical purpose to qualify as an IVD. This is stated in the definition of a medical device according to MDR, Article 2, No. 1, because every IVD is also a medical device as stated in the definition of an IVD in Article 2 No. 2 of the IVDR. In short, in vitro diagnostic medical devices are, therefore, a subgroup of medical devices that obtain medical or diagnostic information from human samples.
You can read more about the qualification of IVDs in this article.
Another article deals with software qualification as IVD medical device software (IVD MDSW).
We take a look at the differentiation from “Research Use Only” products in our article Laboratory products for “Research Use Only” (RUO) – often a dangerous claim.
If you would like to learn more about the distinction between IVD and general laboratory equipment, we recommend our article General laboratory equipment: What manufacturers and laboratories need to know to avoid problems and unnecessary expense.
b) Classifying the device according to IVDR Annex VIII
The IVDR distinguishes between four classes of IVD: A, B, C, and D. The devices with the lowest risk fall into class A, and those with the highest risk fall into class D.
This article describes how IVD manufacturers must classify their devices.
Another article deals with the special features of classifying IVD software.
c) Select conformity assessment procedure
Annexes IX, X, and XI of the IVDR describe the conformity assessment procedures that manufacturers may choose depending on the risk class of the device.
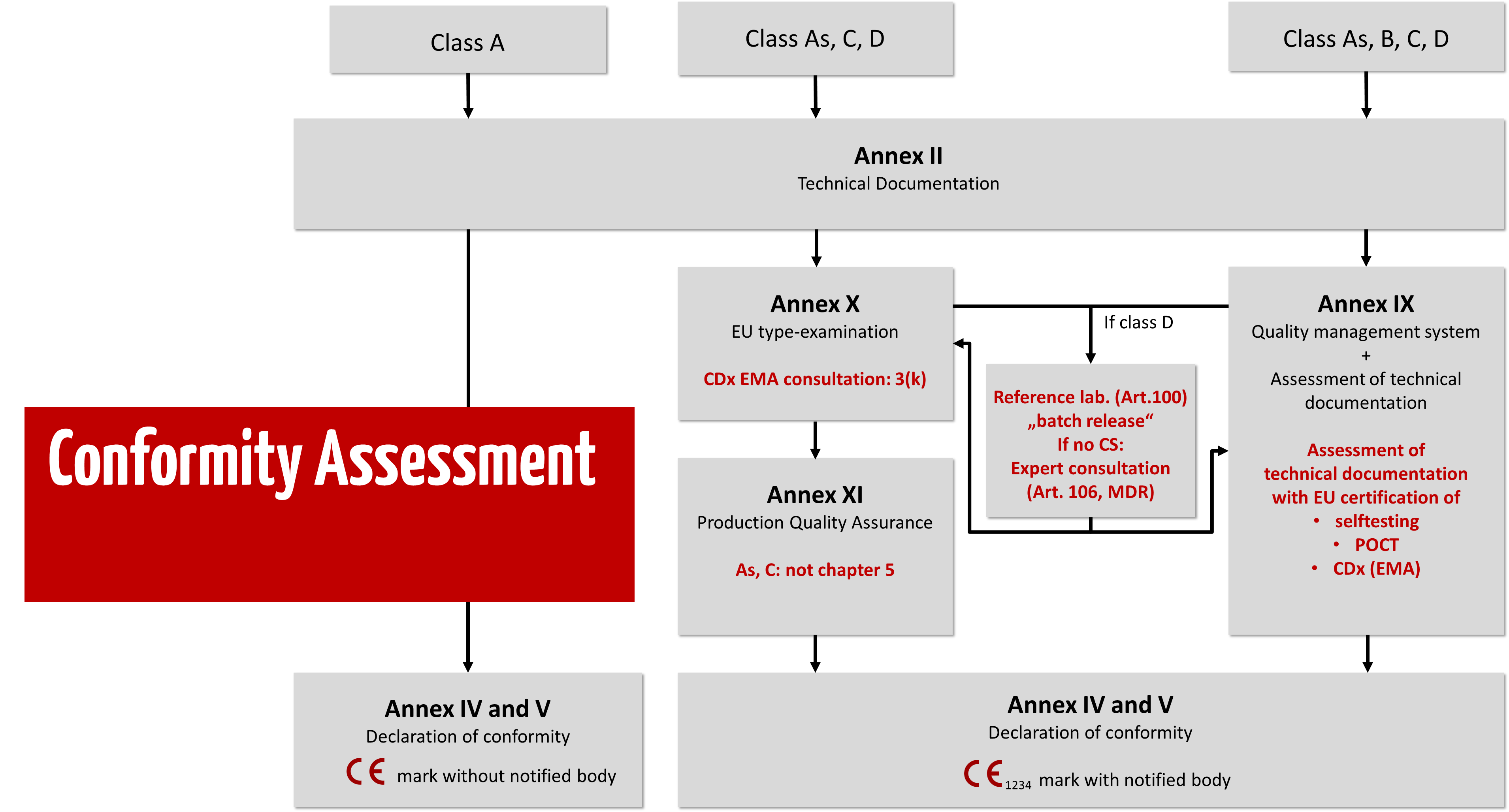
d) Demonstrating applicable safety and performance requirements and creating technical documentation
Regardless of the class and conformity assessment procedure, all IVDs must always meet the general safety and performance requirements set out in Annex I of the IVDR, and the manufacturer must draw up technical documentation in accordance with Annexes II and III of the IVDR.
In-house IVDs must also meet the general safety and performance requirements in Annex I of the IVDR.
Manufacturers usually demonstrate compliance with the general safety and performance requirements based on harmonized standards. Depending on the product type (e.g., reagent kit, instrument, software, specimen receptable), the following standards are relevant for in-vitro diagnostics:
- ISO 13485:2016: Medical devices – Quality management systems – Requirements for regulatory purposes
- ISO 14971: Medical devices – Application of risk management to medical devices
- IEC 62366-1: Usability
- IEC 62304: Software
- ISO 20916: Clinical performance studies with IVDs
- EN ISO 17511: Requirements for the determination of metrological traceability of values assigned to calibrators, trueness control materials, and human samples
- ISO 18113 (Parts 1 to 5): Requirements for information to be provided (symbols, instructions for use, labeling, etc.)
- ISO 23640: Stability testing of reagents for in vitro diagnostic tests
- EN 13641: Elimination or reduction of the risk of infection posed by reagents for in vitro diagnostic tests
- Standards related to sterility: EN 556 (parts 1 and 2), ISO 11737 (part 2), ISO 13408 (parts 1 to 6), and ISO 14937
- IEC 61010-1, IEC 61010-2-101, IEC 61326-1, IEC 61326-2-6: Requirements for IVD devices and instruments
- IEC 61310-1: Alarms
- ISO 11073: Data transmission of point of care devices (not harmonized)
Other product-specific standards exist, such as ISO 15197 for blood glucose meters or EN 14254, which formulates the requirements for materials, nominal filling quantity, graduation, design, execution, sterility, additives, and information by the manufacturer of single-use sample containers, except for blood samples. EN 14820 applies to disposable vessels for venous blood sampling in humans.
In addition to the standards, manufacturers must comply with Common Specifications if available for their specific IVD.
Finally, there are numerous guidance documents, e.g., the CLSI Guidances, which are recognized as state of the art and should be considered by IVD manufacturers, e.g., during performance evaluation.
Read more about the verification and validation of IVDs. (German)
In this article, we have summarized important information on the performance evaluation of in vitro diagnostics: 8 steps to conformity.
If you are unsure which regulatory requirements apply to your IVD or how you can justify non-applicability with an objective rationale, please contact us.
2. Requirements for the approval of special IVDs
a) Requirements for IVD devices and instruments

In addition to the IVD Regulation (IVDR), some other European directives and regulations may be relevant and whose applicability to devices and instruments must be checked by IVD manufacturers:
- 2001/65/EU (Restriction of certain Hazard Substances RoHS) for the avoidance of hazardous substances in electrical and electronic equipment
- 2002/96/EC or the implementing Electrical and Electronic Equipment Act (ElektroG) on the handling of electrical waste
- 1999/5/EC (Radio Equipment and Telecommunications Terminal Equipment, RTTE) on radio equipment and telecommunications, which is also explicitly applicable to medical devices. The successor regulation is 2014/53/EU.
Special case: Machinery Directive or Regulation (EU) 2023/1230 on machinery
Some IVDs have moving parts, and users can be injured if there are no guards. For example, some laboratory machines have “gripper arms.” The Machinery Directive (Directive 2006/42/EC) and the new EU Regulation 2023/1230 on machinery formulate requirements for such devices.
For manufacturers, the question arises as to whether the IVDR, the Machinery Directive, or the Machinery Regulation should be applied. According to Article 51 of the Machinery Regulation (EU) 2023/1230, “Directive 2006/42/EC [will] not be repealed until January 14, 2027“. In addition, the transitional periods set out in Article 52 of the Regulation apply.
Regarding the application of IVDR and/or Machinery Directive, Article 3 of the Directive states:
“Where, for machinery, the hazards referred to in Annex I are wholly or partly covered more specifically by other Community Directives, this Directive shall not apply, or shall cease to apply, to that machinery in respect of such hazards from the date of implementation of those other Directives.”
Directive 2006/42/EC, Article 3
The Machinery Directive formulates it similarly in Article 9:
“Where, for a certain product within the scope of this Regulation, the risks addressed by the essential health and safety requirements set out in Annex III are wholly or partly covered by Union harmonisation legislation that is more specific than this Regulation, this Regulation shall not apply to that product to the extent that that specific Union legislation covers such risks.”
Regulation (EU) 2023/1230, Article 9
This means: If the IVDR formulates the requirements more precisely than the Machinery Directive or Machinery Regulation, (only) the IVDR must be considered for these requirements; for the other requirements, the Machinery Directive or Machinery Regulation must also be taken into account in addition to the IVDR.
Create a table whose first column contains the “union” of the requirements of the IVDR and the Machinery Directive or Machinery Ordinance. In the second column, note whether the requirements affect you and, if so, which of the two contains the more specific requirements. In the third column, note the reference to the implementation (e.g., a product requirement) or its verification.
b) Requirements for operators of IVDs and medical laboratories with developed in-house IVDs
The German Medical Association’s guideline for the quality assurance of laboratory medical examinations (RiliBÄK) is not aimed at manufacturers but at IVD operators. In addition, ISO 15189 determines requirements for the quality and competence of medical laboratories that carry out in vitro diagnostic tests.
Laboratories should avoid becoming manufacturers themselves. The IVDR describes EU-wide regulatory requirements for “devices manufactured and used only within health institutions established in the Union,” so-called in-house IVDs (also known as Laboratory Developed Tests, LDTs) (see IVDR, Article 5 (5)).
Please read our detailed article The EU regulates medical laboratories – Are Laboratory Developed Tests still allowed? to learn more about the IVDR requirements for in-house IVDs.
Our IVD team will happily support you in complying with the IVDR requirements for your in-house IVD.
c) Requirements for sampling kits
According to the definition of an IVD (IVDR, Article 2, No. 2), specimen receptacles are also considered IVDs.
‘Specimen receptacle’ means a device, whether of a vacuum-type or not, specifically intended by its manufacturer for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination
IVDR, Article 2, No. 3
We explain the regulatory requirements for sampling kits in this article.
d) Devices for self-testing and near-patient tests
In the case of IVDs for self-testing and near-patient tests (also known as Point-Of-Care Tests (POCTs)), the focus is on the specific context of use and the user.
‘device for self-testing’ means any device intended by the manufacturer to be used by lay persons, including devices used for testing services offered to lay persons by means of information society services
IVDR, Article 2, No. 5
‘for near-patient testing’ means any device that is not intended for self-testing but is intended to perform testing outside a laboratory environment, generally near to, or at the side of, the patient by a health professional
You can read about the particular regulatory requirements in the article Self-tests and near-patient tests: What EU law says. (German)
3. Conclusion and summary
In Europe, the approval of IVDs under the IVDR is very similar to the approval of medical devices under the MDR.
An appropriate approval strategy based on a precisely formulated intended purpose is crucial for the success of IVD manufacturers. It should provide answers to questions such as:
- Is it a more complex system, and what product components does it consist of (e.g., assay, device, software)?
- How can the system be divided into IVD, accessories, and non-IVD to achieve the simplest possible “approval” and the greatest possible flexibility in use and further development?
- Which classes do IVDs and accessories fall into?
- Which “claims” or performance requirements can and must be made or avoided for the device?
- What is the quickest and safest way to verify these claims during performance evaluation?
Please contact us if you would like the support of the Johner Institute’s IVD experts in
- formulating the intended purpose,
- determining the approval strategy,
- deriving the performance evaluation strategy, or
- creating product-specific documents to demonstrate the general safety and performance requirements.
Change history
- 2024-03-07: Article completely revised
- 2023-04-22: Article revised; references to the IVDD removed
- 2020-09-11: Requirements of the IVDR added


