With the Pre-Submission Program (“pre-sub” for short), the FDA offers a formal procedure for manufacturers to clarify their regulatory strategy and specific questions before actual approval or market clearance. A pre-sub request is suitable in the preparation of 510(k)s, De Novo Requests, or PMAs, among other things. This can avoid unnecessary costs and effort on both sides.
The Johner Institute has observed that many manufacturers are unaware of this offer and/or do not prepare carefully enough for FDA pre-submission meetings.
In this article, you will learn how to get in touch with the FDA and make the most of the Pre-Submission Program – for example, to shorten the review time of your submission.
1. Background: The Q-Submission Program
Pre-submissions allow manufacturers to contact the FDA and obtain feedback as part of the Q-Submission Program. With the help of a pre-submission request, specific questions can be discussed in advance of device approval.
The FDA distinguishes between different Q-submission types:
- Pre-Submissions
- Submission Issue Requests (SIR)
- Study Risk Determinations (SRD)
- Informal Meetings
- Other Types: These Q-Submissions are suitable, e.g., in connection with
- Breakthrough Devices,
- PMA,
- classification of accessories, or
- clinical investigations.
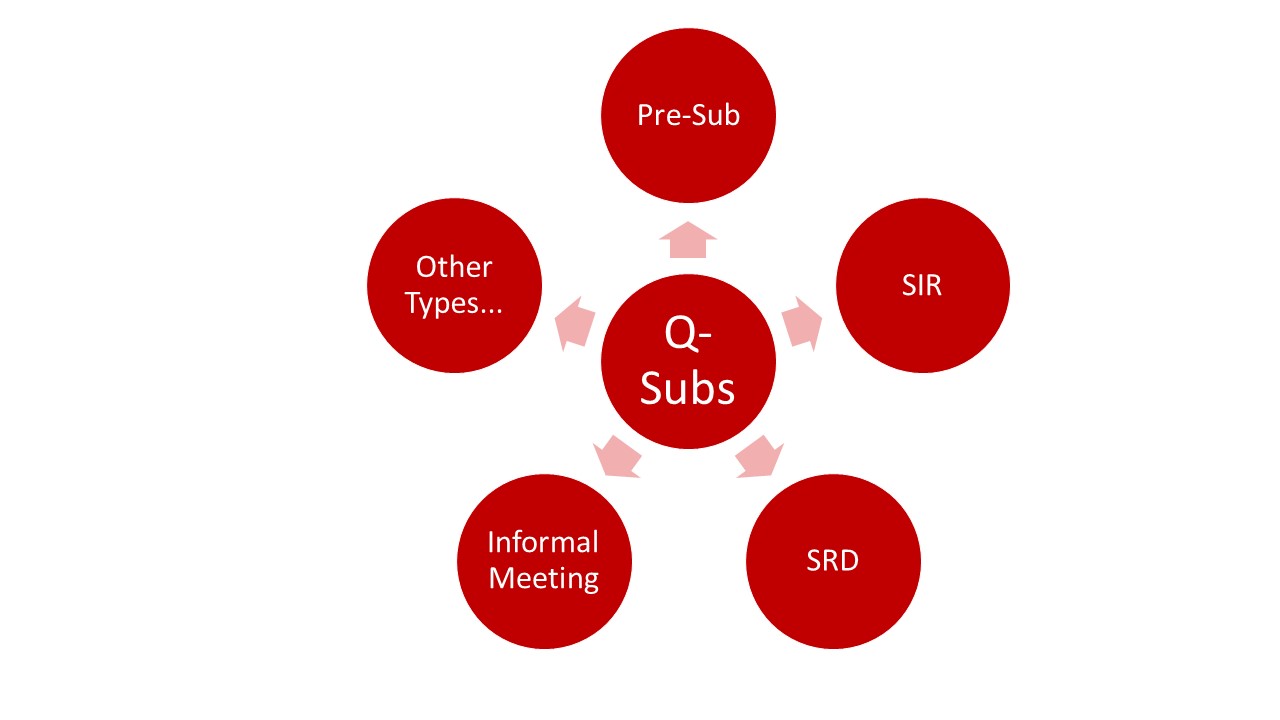
The following article refers to the possibilities of pre-submission requests.
2. When to go through a pre-sub and when not to
The FDA allows manufacturers to submit a pre-submission request in relation to all forms of market approval:
- IDE – Investigational Device Exemption
- 510(k) PMN – Premarket Notification (there are several 510(k) variants)
- PMA – Premarket Approval
- HDE – Humanitarian Device Exemption
- De Novo Request
- CLIA – Clinical Laboratory Improvement Amendments
- Breakthrough Devices
You can submit a pre-submission request if you have specific questions before the submission and would like to coordinate with the FDA. Annex 2 of the associated guidance document contains sample questions that manufacturers can use as a guide. The program is voluntary and free of charge.
a) When the FDA recommends a pre-submission request
A pre-submission request is particularly recommended for the following questions:
- We are planning a De Novo Request. Does the FDA agree that there are no suitable predicate devices available?
- Does the FDA consider the predicate device to be sufficiently equivalent?
- Does the FDA agree that a clinical study is unnecessary based on our regulatory strategy and preclinical data?
- Does the FDA agree with the planned study design?
- Does the FDA consider the planned usability study appropriate for collecting evidence on the safe use of our device?
- Is the software’s documentation level correctly classified?
- Is the cybersecurity management plan sufficient for a planned submission? If not, can the FDA provide us with the missing information?
- Based on the documents submitted, does the FDA consider our Predetermined Change Control Plan (PCCP) appropriate?
Manufacturers should use pre-subs, especially if the regulatory strategy is unclear, when using new technologies, or for “first-of-kind devices.” This also applies if regulations are not entirely clear or not precisely applicable.
b) When the FDA rejects a pre-submission request
The FDA does not want to waste time and rejects pre-sub requests in the following cases, for example:
- The manufacturer does not ask specific questions.
- The answer could also be given directly by an FDA reviewer.
- It is (only) about the classification of the device. In this case, manufacturers must submit a 513(g) application.
- The request corresponds to a different type of Q-submission.
3. Content of a pre-sub request
The FDA has precisely defined the contents of a pre-submission request. This way, it wants to ensure the process is efficient and suitable for all parties involved. The associated guidance document is called Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program and requires the following content:
- Cover letter
- Type of Q-submission request
- Contact details of the manufacturer (sponsor)
- Name of the medical device
- Reason for requesting feedback
- CDRH Premarket Review Submission Cover Sheet (Form FDA 3514)
- Table of contents
- Detailed description of the medical device, including intended purpose
- History of previous approvals of the device or earlier communications with the FDA
- Overview of the product development, including (planned) test strategy
- List of questions for which answers are desired
- Desired communication channel (in person, in writing, by telephone)
- Three suggested dates for the meeting or telephone call
- Planned participants
4. Typical sequence of the pre-submission procedure
A pre-submission is a formalized procedure. The FDA answers the manufacturer’s questions
- during a telephone conference (preferred by the FDA),
- as a written response, or
- during a personal meeting (“face to face meeting”).
The pre-submission procedure is also clearly defined (see Fig. 2):
- The manufacturer drafts and submits the pre-submission to the FDA in eCopy or eSTAR format. This includes a cover letter and the CDRH Premarket Review Submission Cover Sheet. The cover letter contains all the information the FDA needs to answer the manufacturer’s questions and, if necessary, schedule a meeting.
- The FDA responds within 15 days and determines whether the manufacturer has selected the correct Q-submission type and whether all necessary information is available. If not, the FDA will request the missing information or reject the pre-submission. This review by the FDA is called RTA (Refuse to Accept). The FDA uses the checklist in Annex 1 of the guidance document for this purpose. If necessary, the manufacturer submits information as an “amendment.” Only when the manufacturer has sent this missing information, the FDA starts the 70-day period.
- Both parties agree on the meeting, which typically takes place 60 to 75 days after receipt of the pre-submission.
- The FDA sends its preliminary responses at least five days before the meeting. Any presentation documents must be emailed to the FDA at least two days before the meeting.
- The meeting takes place (usually for one hour; more time requires justification). The manufacturer must document the meeting and forward the meeting notes to the FDA.
- The manufacturer sends the meeting notes to the FDA to amend the original pre-submission in eCopy format within 15 days.
- If necessary, the FDA corrects these notes (within 30 days) if its understanding of what was written does not match that of the manufacturer. Otherwise, it accepts the notes by email.
- If the manufacturer does not object within 15 days, the meeting protocol is “official.” Otherwise, he objects with a “Meeting Minutes Disagreement.”
- After 75 to 90 days, the manufacturer has the FDA’s feedback, which may contain further comments and concerns from the authority.
Q-submissions must be submitted in accordance with the eCopy Program or according to eSTAR (using the PreSTAR template). The FDA will only begin processing a submission that meets the eCopy requirements or when having received a valid PreSTAR.
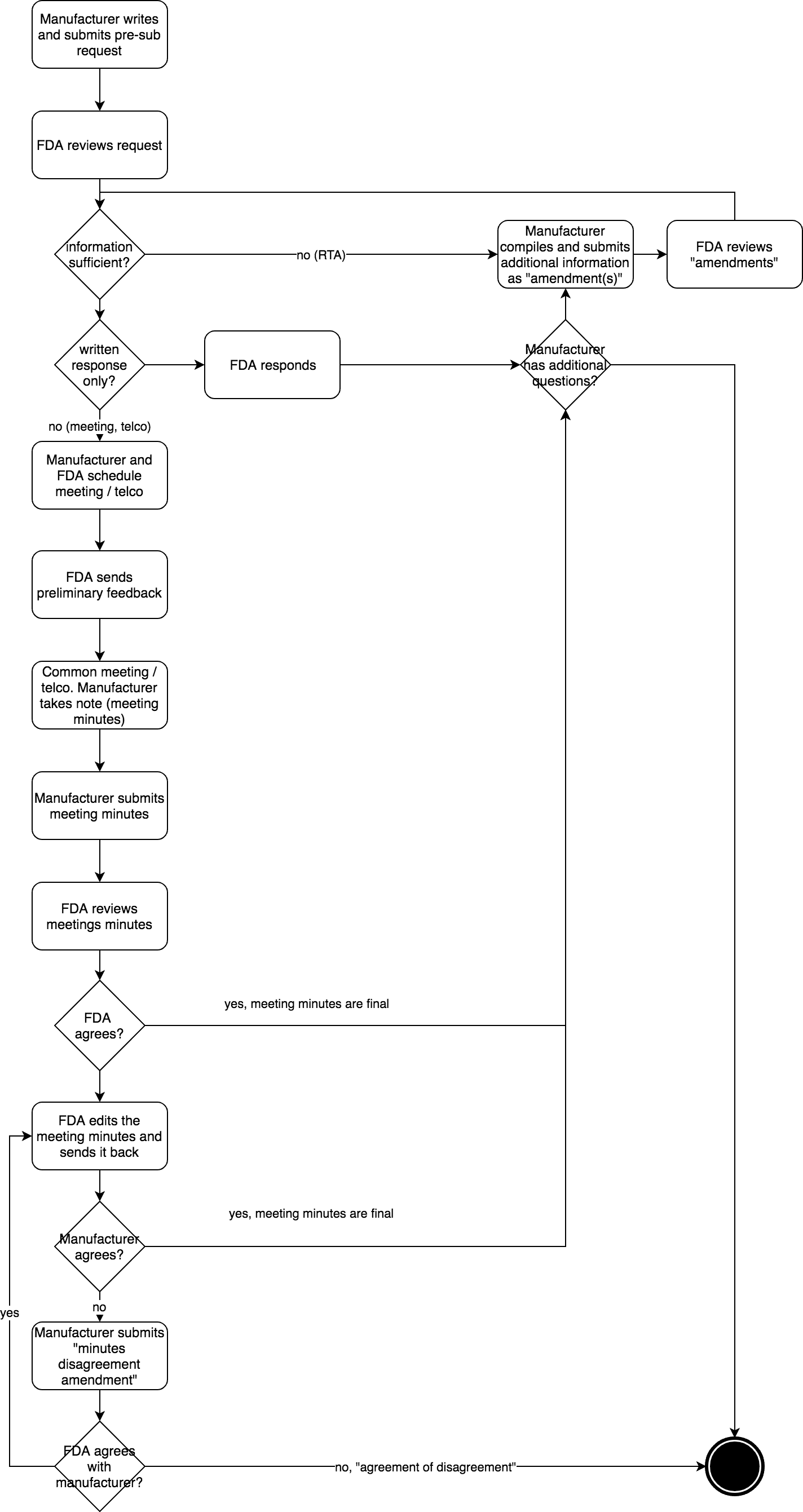
Manufacturers are allowed to add documents during the procedure. These “amendments” bear the “Q-number” and the post-fix “/A001,” “/A002,” etc. If the manufacturer asks additional questions, this is done in the form of “supplements” with the post-fix “/S001.”
However, if the manufacturer substantially changes the device (especially its intended purpose) or if the Q-submission procedure itself is changed (e.g., from pre-sub to “Determination Meeting”), the FDA assigns a new Q-number.
5. Mistakes you should avoid at pre-submission meetings
The Johner Institute repeatedly observes mistakes made by manufacturers in pre-submissions. You should avoid these at all costs to avoid causing yourself more harm than benefit with the pre-subs:
a) Lack of clarity about your own objectives
At the beginning of any process, the objectives should be clear. In the case of a pre-submission request, manufacturers must formulate their questions with absolute precision. Only then can the FDA answer just as precisely. And only then, it is clear what information is necessary to provide the answers.
The FDA does not appreciate manufacturers iteratively “working their way up” to the actual question and providing data in slices. Instead, it wants to work through a topic within one iteration.
However, the FDA allows additional questions to be added within the same procedure (identical Q-number).
b) Unclear strategy
It is essential that manufacturers consistently pursue a clear and easily understandable line of reasoning. Otherwise, they leave it up to the FDA to figure things out, which doesn’t always go well.
Manufacturers should know before the meeting what tests and clinical investigations they intend to conduct and why they believe these are sufficient. Otherwise, the FDA will “persuade” them to conduct more tests, which greatly impacts timelines and costs.
An intelligent strategy also includes the correct form of interaction with the FDA. The Johner Institute recommends in-person meetings (in Silver Spring) for De Novo procedures, PMAs, if available, and conference calls in many other cases.
c) Poor preparation and coordination
Probably the most common mistake is inadequate preparation. The entire procedure must be precisely prepared:
It must not happen that information, or even the manufacturer’s experts are missing at the pre-submission meeting, and then not all questions can be answered (e.g., the clinicians ask questions about planned studies, or the developers ask questions about the technology). It is even worse if the experts contradict each other in the meeting.
A meeting needs to be practiced – almost like a play, where all actors know what to say and what not to say at all times.
If the manufacturers spend most of the time at the meeting with their presentations, they should not be surprised if there is no time left for the FDA’s answers. The agenda is, therefore, an essential element of preparation.
It is also a sign of good preparation if the manufacturer organizes a person whose sole task is to take meeting minutes. Audio or even video recordings are prohibited.
d) No prompt action
Some manufacturers seem so relieved or exhausted after pre-submission meetings that they fail to meet deadlines. In addition, it is very important to document the meeting while everything is still at the top of your mind – even with the FDA.
e) Formal mistakes
The FDA only wants to answer specific questions in pre-submission requests. Classifications are not included. That is the objective of a 513(g) request. Questions that a reviewer can answer directly also have no place in a pre-submission.
Manufacturers should be aware of the different types of Q-submissions as well as other procedures, such as non-formal inquiries and the 3rd party review program. However, the latter is not possible for all medical devices and approval procedures.
6. Conclusion
With its Pre-Submission Program, the FDA offers manufacturers an effective and efficient procedure for obtaining rapid yet binding feedback on questions relating to the approval of medical devices.
The authority itself recommends the procedure for good reason. Manufacturers should also use it to minimize approval risks and delays and avoid unnecessary expenses, especially for studies (including those without an IDE or outside the US).
Of course, manufacturers should only submit a pre-submission request if they have questions. Otherwise, the procedure may even delay approval.
The entire procedure must be prepared carefully. Even though we are speaking “pro patria” here: Consider having a consultant help you prepare for and conduct pre-submission meetings. It’s not a big deal, but it will help speed up the process and make it safer. Don’t hesitate to contact us.
Change history:
- 2024-10-16: Adaptation to the current edition of the Q-Sub Guidance from 2023
- 2021-10-08: Adaptation to the current edition of the Q-Sub Guidance from 2021
- 2020-11-06: Revision of the article structure, insertion of current references, updates based on the guidance document from May 2019


