Deviations, nonconformities, errors, findings, observations, and other terms are often used mistakenly synonymously. Even standards explicitly contradict each other when defining individual terms.
This article clarifies,
- how the terms are to be understood,
- what the causes and consequences of nonconformities are, and
- how medical device manufacturers can avoid these deviations and nonconformities.
1. Nonconformities: The basics
1.1 Definitions
1.1.1 Nonconformity
ISO 9000:2015 defines nonconformity as “non-fulfilment of a requirement.” It equates this term with “error.” ISO 13485 contradicts this.
This standard intentionally uses the term “nonconformity” and not the term “error”. The terms are not identical in meaning. “Error” is a legal term that plays a role in a warranty for defects and product liability. Nonconformity differs from this term in that it refers to manufacturer’s own specifications not adhering to those agreed upon with the customer or those expected by the market.
Source: EN ISO 13485:2016 + AC:2018 + A11:2021
The FDA defines the term in a similar way to ISO 9000:
Nonconformity means the nonfulfillment of a specified requirement.
Source: 21 CFR part 820.3(q)
1.1.2 Deviation
Neither ISO 9000 nor ISO 13485 define the term “deviation,” which is often used colloquially as a synonym for nonconformity.
The term originates more from the automotive sector around IATF 16949, which also uses the term “nonconformity.” However, the supplementary set of rules (Rules for achieving and maintaining IATF Recognition IATF Rules 5th Edition) in German also uses the term “deviation” (“Abweichung”) and distinguishes between major and minor deviations.
In the medical device ecosystem, these major and minor deviations are equated with major nonconformities and minor nonconformities in audits.
In English, the term “nonconformity” is used consistently.
1.1.3 Findings
You regularly hear the statement that there was a finding during an audit. This refers to a nonconformity. This colloquial usage does not match the definition of the term:
results of the evaluation of the collected audit evidence against audit criteria.
Note 1 to entry: Audit findings indicate conformity (3.20) or nonconformity (3.21)
Note 2 to entry: Audit findings can lead to the identification of risks, opportunities for improvement or recording good practices.
Note 3 to entry: In English if the audit criteria are selected from statutory requirements or regulatory requirements, the audit finding is termed compliance or non-compliance.
Source: ISO 19011:2018, 3.10 (ISO 9000:2015, 3.13.9, modified)
Audit findings, i.e., “outputs of the assessment of the compiled audit evidence against audit criteria,” are therefore inevitable in audits. However, this does not mean that findings are automatically negative and should be avoided. The situation is different for deviations or nonconformities.
1.1.4 Defect
The term “defect” should also not be equated with the term nonconformity.
“The distinction between the concepts defect and nonconformity is important as it has legal connotations, particularly those associated with product (3.7.6) and service (3.7.7) liability issues.”
Source: ISO 9000:2015
1.1.5 Further terms
Even the FDA is not entirely consistent. It uses other terms such as deviation, nonconformance, observation, and violation in the context of inspections without defining them. Examples of this can be found in the Compliance Program Guidance Manual, Inspection of Medical Device Manufacturers, the QSIT, and the Investigations Operations Manual.
1.2 Examples for nonconformities
Nonconformities can be found in both devices and processes.
1.2.1 Non-conforming processes
Examples of non-conforming processes are
- Processes that are not implemented at all or are not carried out in accordance with documented standard operating procedures.
Example: No assessment of employee competence has taken place. - Processes that are not defined.
Example: There is no process for evaluating and remedying nonconformities. - Processes that do not meet the requirements of a standard or legal requirement.
Example: The standard operating procedure does not require complaints to be assessed to determine whether a complaint constitutes a reportable incident.
These nonconformities are typically identified during audits and inspections.
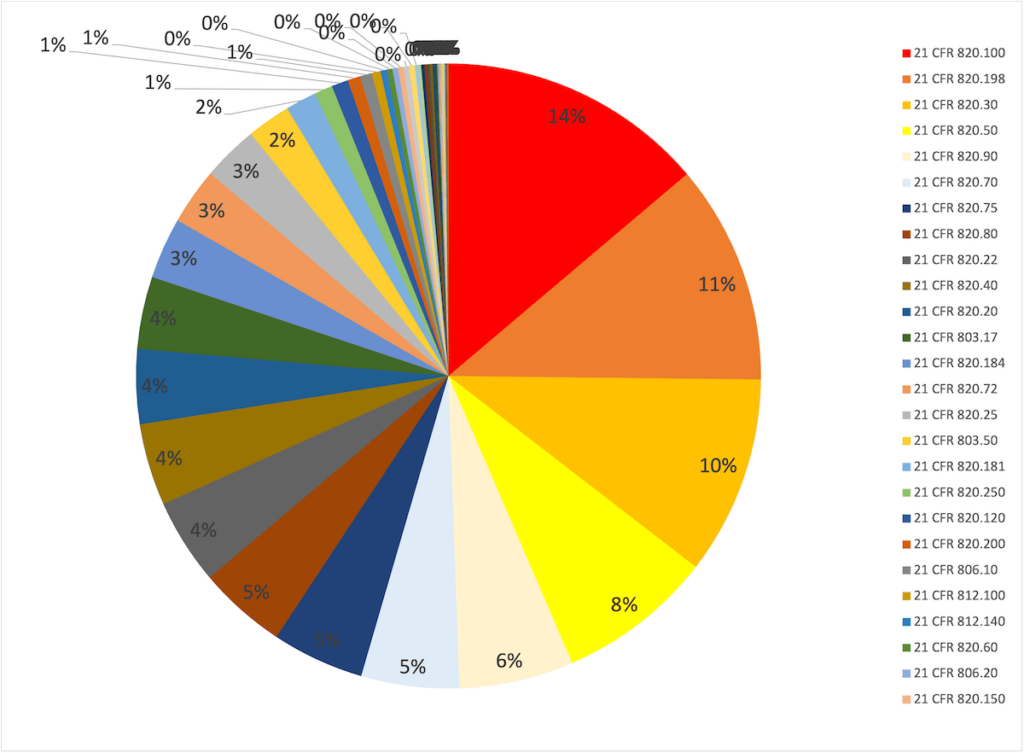
Nearly half of the nonconformities the FDA found during inspections in 2023 are:
| regulatory requirement | description | share |
| 21 CFR 820.100 | corrective and preventive actions | 14 % |
| 21 CFR 820.198 | complaint handling | 11 % |
| 21 CFR 820.30 | design control | 10 % |
| 21 CFR 820.50 | supplier | 8 % |
| 21 CFR 820.90 | Handling of non-conforming products | 6 % |
| rest | 51 % |
1.2.2 Non-conforming products
Examples of non-conforming products are
- Devices whose usability has not been demonstrated
- Medical electrical equipment with excessive leakage currents
- Devices that do not meet the advertised specification
- Devices whose medical benefit has not been proven and/or for which this benefit does not outweigh the risks
- Instruments that release toxic substances into the body
2. Consequences of nonconformities
The consequences of nonconformities depend on the possible consequences of these nonconformities:
- Seizure or/and disposal of the devices by the authorities
- Obligation to recall the devices by the manufacturer
- Withdrawal of certificate
- Warning letters
- Criminal prosecution
- Import ban
- Inspection by authorities
- Fines
- Obligation to rectify nonconformities and their causes (corrective actions)
For example, the German MPDG gives the authority the following rights:
In particular, the competent authority is authorized within the scope of this Act
1. to prohibit or restrict the placing on the market or putting into service of the device,
2. to prohibit or restrict the making available on the market of a device,
3. to order measures to ensure that a device is only placed on the market or made available on the market if suitable and easily understandable safety instructions are included in the label or in the instructions for use,
4. order the withdrawal or recall of a device made available on the market,
5. to prohibit or restrict the operation or use of the device concerned,
6. order that the public be warned of the risks posed by a device made available on the market; the competent authority may itself warn the public if the economic operator fails to warn or fails to warn in good time or fails to take other equally effective action in good time.
MPDG § 74, Procedures for protection against risks, Section 2 (translated from German)
The MDR authorizes the authorities to take the following measures:
The competent authorities may confiscate, destroy or otherwise render inoperable devices that present an unacceptable risk or falsified devices where they deem it necessary to do so in the interests of the protection of public health.
MDR, Article 93, Part 5
3. Causes of deviations or nonconformities
The causes are varied. However, they can usually be traced back to one of the following reasons:
- Those responsible are not aware of the legal requirements and requirements of applicable standards.
- The legal and normative requirements were not correctly understood and implemented.
- The legal and normative requirements were knowingly ignored.
- The company’s own internal requirements are too complicated.
- The management has not communicated the importance and meaning of the requirements to the employees.
4. Finding and avoiding nonconformities
4.1 Finding nonconformities
Manufacturers should look for nonconformities themselves by:
- Carrying out internal audits (mandatory under ISO 13485)
- Carrying out tests to find nonconformities, possibly also automated
- Inspections and reviews of devices and documents
Manufacturers can call on external experts for these inspections, such as internal audits and mock inspections.
The experts at the Johner Institute carry out these audits and mock inspections. They not only identify possible nonconformities but also provide concrete assistance to eliminate and avoid them in the future.
4.2. Avoiding nonconformities
Comprehensive quality management focuses not only on finding nonconformities but also on avoiding them.
- Manufacturers should design their processes in such a way that they are understandable, sensible, and lean. In this way, nonconformities can be avoided from the beginning.
- Manufacturers can call on external expertise to explain the requirements and their interpretation to investigators and authorities.
- Manufacturers should use methods/tools to avoid nonconformities, such as automated workflows, electronic forms with inspection, training systems, and documentation of employee training.
5. Conclusion and summary
The term deviation is only used colloquially. It is, therefore, advisable to speak of nonconformities, even if the FDA uses different terms, mixes them up, and does not define them.
Whether deviation or nonconformity, manufacturers must avoid or at least identify and eliminate these deficiencies. This is because the consequences for both manufacturers and individuals are far-reaching and include criminal prosecution and the end of the company.
The Johner Institute’s remediation service assists manufacturers whose authority or notified bodies have identified nonconformities and who are threatened with consequences.

{kind=link}
Dear Luca,
Thank you for this comprehensive article. I like it a lot, because this terminology raised often questions. However, I do not agree completely, i.e. I would like to proceed using the term “Deviation” for flexibility and and efficiency purposes in the daily operations within a well implemented and conform QMS.
For me, a Deviation is a “controlled different behavior from the originally defined and agreed upon way of working (SOP).” This compared to a non-Conformity which is, as you indicated, most of the time detected during Reviews, Audits and Inspections. Detected by “someone else, they seem to be “un-controlled.”
For practical reasons, in daily practice, it might be necessary to deviate from the normal in order to avoid interruption of the workflow. Let’s say, during the preparation of a solution, according to the Work Instruction, a particular scale needs to be used. But the scale is not available due to repair. Another scale is available and, according to the logbook, suitable and available, The alternative scale is used for the preparation of the solution, this Deviation of the WI is recorded in the Batch Record, the related QC shows satisfying results, which means that this non-Conform activity does not harm the product.
Another example: it is mandatory that a particular person signs a document but is not available and is not able to sign and approve that document. The sign-off cannot wait and an alternative is not (yet) defined in the SOP. Therefore, the situation is discussed by phone, the review performed “by app” and a oral approval has been communicated. Somebody else, with who the responsible for the signature in in contact, signs the document p.o. on the date reviewed and approved and a formal sign-off by the right person is organized the moment he is in person recording the date of his signature (two signatures, two dates, and a description of the Deviation documented.
According to clause 8.2.5 of EN-ISO 13485:2016 the processes of the QMS are monitored and measured periodically, the Deviation is found (because documented immediately when it occurred) analyzed, reassessed, and reported in the MR providing an opportunity to resolve the issue when it occurs repeatedly (update the documented procedure) or just report and discuss it when it seems to be an incident.
All well within an controlled environment, nicely documented ad assessed on a proper moment. It is Deviation, but not a non-Conformity.
Two other remarks on your article:
– I would use EN-ISO 13485:2016+A11:2021 instead of ISO 13485:2021, which does not exist.
– I would mention the applicable definitions as provided in EN-ISO 19011 as well. (Definition Audit Findings: results of the evaluation of the collected Audit Evidence against Audit Criteria. Audit Findings indicate Conformity or non-Conformity. Audit Findings can lead to the identification of Risks, Opportunities for Improvement or recording Good Practices. In English, if the Audit Criteria are selected from Statutory Requirements or Regulatory Requirements, the Audit Finding is termed Compliance or non-Compliance.
SOURCE: ISO 19011:2018, definition 3.10: ISO 9000:2015, 3.13.9, modified – notes to entry 2 and 3 have been modified)
Dear Louis,
thank you very much for your great feedback and for sharing your thoughts on the meaning of “deviation”. This goes more in the direction of MIL-STD-973’s definition:”A specific written authorization, granted prior to the manufacture of an item, to depart from a particular requirement(s)…”.
This is why it is so important to have defined terms, especially in the regulatory field. I saw even Notified Bodies and medical device authorities using the term “nonconformity” and “deviation” interchangeably.
Best regards
Luca
PS: The wrong reference to ISO 13485 has been corrected. We included the modified definition of “audit finding” from ISO 19011.