Companion Diagnostics (also known as CDx) are used together with a medicinal product. Physicians use them, for example, to make sure that a particular medicinal product is actually suitable for a patient. This means that CDx play a particularly important role in personalized medicine.
As the Companion Diagnostic and the medicinal product are inseparably linked, there are some specific features in their development and approval.
In this article you will learn:
- how Companion Diagnostics are defined,
- what the development and approval of CDx looks like in practice,
- what regulatory hurdles must be overcome in Europe and the USA, and
- what tips we have so your CDx development and approval can be successful.
1. What are Companion Diagnostics (CDx)?
The success of some drug therapies requires the use of an in vitro diagnostic medical device (IVD). Laboratories use this IVD to determine whether a biomarker that the drug targets is present in the patient. Physicians can use the analysis from the IVD to make sure that a particular therapy is actually suitable for their patient.
Usually, the CDx detects gene sequence changes in the corresponding analyte or the presence of certain proteins.
Typical platforms are:
- Immunohistochemistry
- Quantitative PCR
- Next-generation sequencing
- In-situ hybridization (FISH, CISH)
If a particular result from the IVD is a prerequisite for a safe drug therapy, then this IVD is called a Companion Diagnostic (CDx). Ideally, CDx are developed in close collaboration with medicinal product manufacturers.
Based on Recital (11) of the IVDR, the main intended uses of CDx are as follows:
- Defining patients’ eligibility for specific treatment with a medicinal product through the quantitative or qualitative determination of specific markers.
- Identifying subjects at a higher risk of developing an adverse reaction to the medicinal product in question.
- Identifying patients in the population for whom the therapeutic product has been adequately studied, and found safe and effective. Such biomarker or biomarkers can be present in healthy subjects and/or in patients.
However, the definitions of CDx in Europe and the USA are different.
a) Different definitions of CDx
Europe
In Europe, the term “companion diagnostic” is defined by Regulation 2017/746 on In Vitro Diagnostic Medical Devices (IVDR):
“companion diagnostic” means a device which is essential for the safe and effective use of a corresponding medicinal product to:
- (a) identify, before and/or during treatment, patients who are most likely to benefit from the corresponding medicinal product; or
- (b) identify, before and/or during treatment, patients likely to be at increased risk of serious adverse reactions as a result of treatment with the corresponding medicinal product;”
Source: Art. 2(7) IVDR
Explicitly not CDx
“Devices that are used with a view to monitoring treatment with a medicinal product in order to ensure that the concentration of relevant substances in the human body is within the therapeutic window.”
Recital (12) IVDR
This exception is particularly important because it does not exist in the USA. The FDA’s definition of Companion Diagnostic is different to the European definition:
USA
The FDA defines “IVD companion diagnostic devices” as follows:
“An IVD companion diagnostic device could be essential for the safe and effective use of a corresponding therapeutic product to:
- Identify patients who are most likely to benefit from the therapeutic product
- Identify patients likely to be at increased risk for serious adverse reactions as a result of treatment with the therapeutic product
- Monitor response to treatment with the therapeutic product for the purpose of adjusting treatment (e.g., schedule, dose, discontinuation) to achieve improved safety or effectiveness
- Identify patients in the population for whom the therapeutic product has been adequately studied, and found safe and effective, i.e., there is insufficient information about the safety and effectiveness of the therapeutic product in any other population”
Source: FDA
Manufacturers who want to place their in vitro diagnostic medical device for monitoring a drug therapy on the market in Europe and the USA should make sure they are aware of these different definitions.
b) Differentiation from complementary diagnostics
There is another group of IVDs that are not Companion Diagnostics but that could be confused with them: “complementary diagnostics.”
Complementary diagnostics are neither defined nor described in the IVDR. Their use is recommended in combination with a specific medicinal product, rather than being mandatory before the medicinal product can be applied. Therefore, they are used only to help decision-making, not as a required part of the therapy.
This means complementary diagnostics do not have a close link with a drug in the way that Companion Diagnostics (CDx) do. As a result, from a regulatory perspective, complementary diagnostics are treated in the same way as “traditional IVDs”, i.e., the specific features that apply to CDx do not apply.
Example:
An FDA-approved complementary diagnostic is an immunohistochemical assay for the detection of PD-L1 proteins in patients with non-small cell lung cancer who are candidates for cancer immunotherapy (Nivolumab).
This is a complementary diagnostic and not a CDx because a therapeutic benefit has been demonstrated for ALL patients who receive the medicinal product irrespective of their biomarker status. Nevertheless, there are some patients who respond better to the medicinal product than others: patients who express particularly high levels of the protein PD-L1.
Therefore, the use of the IVD is not mandatory, but does provide a prognosis for how effective the drug treatment might be.
- Guidance document MDCG 2020-16 rev.2 Annex II includes a flowchart to help determine whether an IVD is a CDx
- Glossary of terms related to Companion Diagnostics (CDx) and complementary diagnostics
- Clinical Evidence Requirements for CE certification under the In Vitro Diagnostic Regulation in the European Union from MedTech Europe
2. CDx in practice
a) Who are CDx relevant for?
- Medicinal product manufacturers
- Medicinal product manufacturers should familiarize themselves with the regulatory basics for IVDs and the interdependencies between the IVD and medicinal product.
- IVD manufacturers
- IVD manufacturers considering or already developing a CDx should familiarize themselves with the specific requirements for the development and approval of CDx. There are higher regulatory hurdles for CDx than for other IVDs.
b) Applications of CDx
CDx play an important role in “personalized medicine”. The aim is to identify a customized therapy for an individual patient or the causes of their disease and, if necessary, to continually adjust it. The classic use of CDx is in oncology. This is represented by the FDA’s List of Cleared or Approved Companion Diagnostic Devices.
One of the first CDx was a system to detect the level of HER-2 (also known as ERBB2) expression in breast cancer. This test is based on an immunohistochemical procedure and is used to decide whether a patient is an eligible candidate for treatment with Herceptin (Trastuzumab). Herceptin is authorized for the treatment of metastatic breast cancer but is not effective in HER-2 negative patients and may even have detrimental effects in these patients. Therefore, the use of a CDx is essential for identifying patients who may benefit from the therapy.
c) What does the process for the co-development of CDx look like?
As CDx and medicinal products are interdependent, the development (and approval) of the two runs in parallel. There are various phases in CDx development that correspond to medicinal product clinical phases. Figure 1 provides an overview. We explain the individual phases in more detail below.
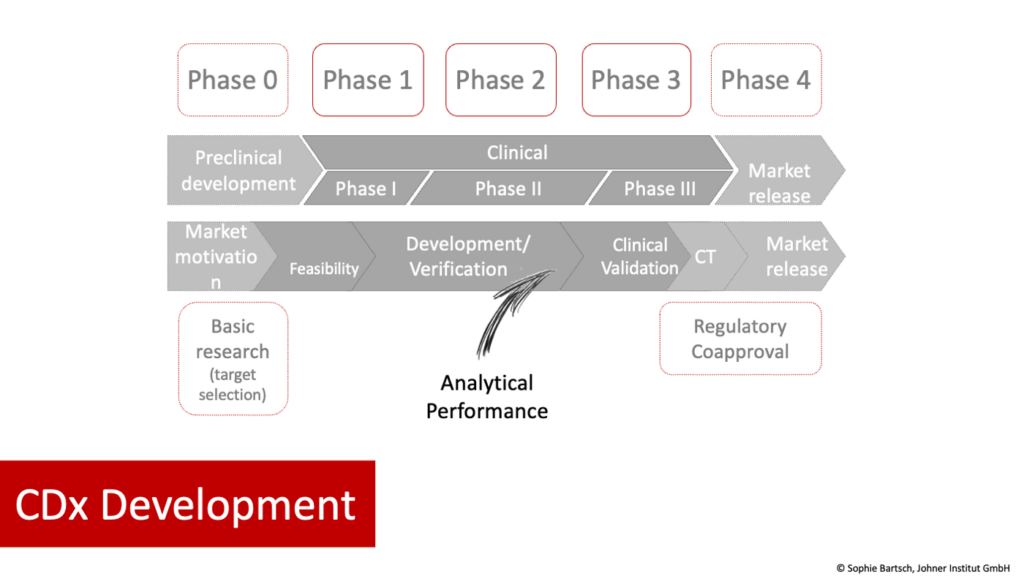
Phase 0:
- Medicinal product
- Preclinical phase
- Answering the questions: What type of diagnostics are needed to correctly identify the patient population for the medicinal product AND to understand an individual patient’s disease process?
- IVD (CDx)
- Basic research/development
- Including selection of analyte(s) and methods. As part of this process, the IVD manufacturer should demonstrate the scientific validity that generates the link between the analyte and the patient’s clinical condition/disease. It should provide the scientific rationale for the use of the biomarker.
Phases 1 to 3
- Medicinal product
- Clinical phases I–II
- Planning and performance of phase III study with the involvement of the CDx
- IVD (CDx)
- While the medicinal product goes through clinical phases I and II, the CDx goes through guided product development and analytical performance evaluation.
- The analytical performance evaluation, i.e., the evaluation of the ability of a device to correctly detect or measure a particular analyte, must be completed before phase III studies with the medicinal product are started in order to ensure well-designed studies.
- This is followed by the clinical performance evaluation of the IVD and clinical investigation of the medicinal product. These are two different studies, with their own protocols and endpoints, that can run in parallel and be aligned with one another. The clinical performance evaluation is used to demonstrate that the IVD is suitable for stratifying or selecting patients for further drug therapy. This enables IVD manufacturers to demonstrate the intended purpose of the IVD in the actual patient population based on data. The clinical study of the medicinal product should show, amongst others, that the patient population identified by the IVD will benefit from the therapy.
Phase 4
- Medicinal product
- Approval
- IVD (CDx)
- Declaration of conformity
- There is no legal requirement for simultaneous approval of the drug and certification of the IVD.
3. Specific regulatory features of CDx
As the purpose of the IVD and the drug are inseparably linked, the regulatory requirements for medicinal products and CDx also intertwine. The specific regulatory requirements for CDx take both the IVD (CDx) and the medicinal product into account.
a) Specific regulatory features in Europe
The IVDR contains separate regulations for CDx. The following aspects are the most significant:
- The regulatory requirements for CDx are stricter than those for other IVDs
- CDx are in risk class C
- Notified bodies and drug authorities work together on a consultation process as part of the conformity assessment
- The clinical evidence follows the same process as for other IVDs: scientific validity, analytical performance, clinical performance. For the latter, the manufacturer also needs studies that show that the CDx is able to stratify or identify patients for a specific therapy based on a particular biomarker.
The specific regulatory features are:
Risk class
Companion Diagnostics are assigned to risk class C in the risk classification according to Annex VIII of the IVDR. However, there are additional regulatory requirements for CDx that go beyond the requirements for other class C IVDs (see below). There are examples in MDCG Document 2020-16.
Special conformity assessment procedures
Normally, the conformity assessment for class C devices is only carried out on one representative device per generic device group. However, it is different for CDx: Art. 48(7) of the IVDR states that each individual CDx device must go through the procedure. This means CDx manufacturers cannot take advantage of device groups.
Consultation with the drug authorities
CDx can be conformity assessed according to Annex IX of the IVDR. However, Section 5.2 of the annex must also be considered.
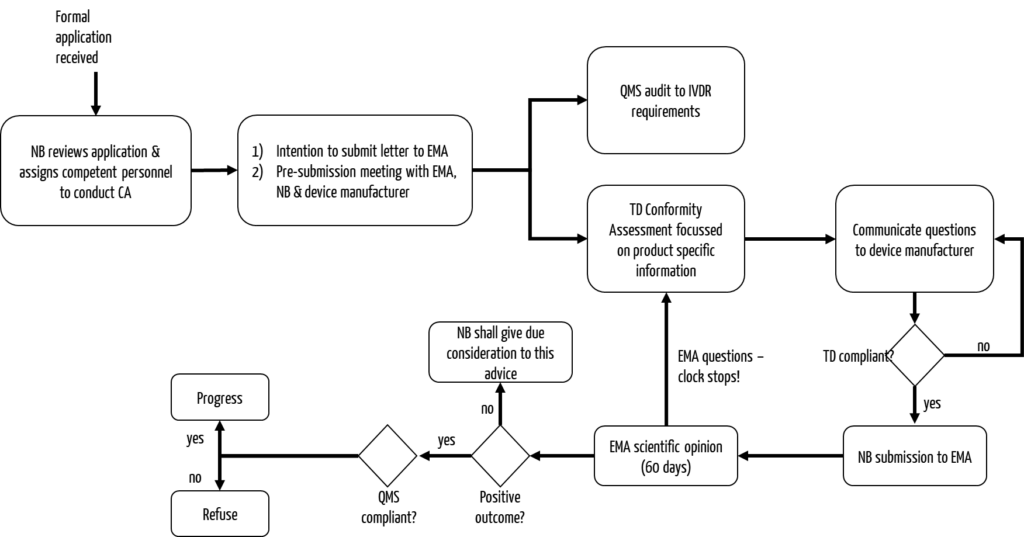
It is particularly noteable that, in addition to the notified body, a drug authority has to be involved. Usually, this is the European Medicines Agency (EMA).
- Role of the drug authority
Annex IX, Section 5.2 describes the requirements for the involvement of a drug authority in the conformity assessment procedure. It states that the drug authority may be either a national competent authority or the EMA. However, new medicinal products are usually reviewed by the EMA.
National authorities may be involved in some cases regarding the authorization of some legacy devices, where necessary. - Process for the involvement of the drug authority
- The notified body informs the EMA about the start of an evaluation procedure of a CDx; this includes the submission of an “Intention to submit Letter” at least three months before the planned submission. (The contents of the Intention to submit Letter can be found here: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics).
- EMA fixes the “rapporteur,” who leads the evaluation of an application and is also responsible for evaluating the companion medicinal product.
- In support, the notified body may send questions to EMA before the scheduled submission of the application. If additional guidance is required, the notified body may request a pre-submission meeting.
- The notified body requests a scientific opinion from the Medicines Agency regarding the suitability of the CDx in combination with the medicinal product. The application form can be downloaded from the EMA website. The applicant is the notified body.
- The scientific opinion is based on the draft instructions for use and the summary report on safety and performance. The evaluation regarding the suitability of the CDx in combination with the medicinal product will consider the scientific rationale of biomarker selection, analytical and clinical performance, clinical safety, and clinical benefit to patients.
- The timeframe for consultation is 60 days, with the possibility of an extension for an additional 60 days.
- Please note: If there are queries by the authority, the timeframe will be extended by the time it takes to address the queries.
- In addition to this initial consultation process, the authority may also need to be involved when changes are made to the CDx. This is the case, for example, if the changes affect the performance and/or intended purpose and/or suitability of the CDx in relation to the drug product. A follow-up consultation process is initiated. In this case, EMA provides its opinion within 30 days of receipt of all necessary documents.
CDx based on genetic testing: No patient counseling
If a CDx is based on genetic testing, patient counseling, which is required for other genetic tests, is not required (Art. 4(3) IVDR).
Performance evaluation
Generally speaking: CDx always require clinical data and fall under the scope of “certain performance studies” referred to Art. 58(2) IVDR.
The three pillars of clinical evidence – scientific validity, analytical performance and clinical performance – naturally also apply to CDx.
- The scientific validity should also clarify the clinical performance of the associated medicinal product in the patient population stratified/selected by the CDx.
- The analytical performance evaluation should be completed before the clinical trial of the medicinal product is started (see also Section 2.c) “Co-development process”).
- Clinical performance studies are required to demonstrate clinical performance.
MedTech Europe has summarized further information on this topic.
Otherwise, there are no special requirements for CDx performance evaluations. Nevertheless, EMA will pay particular attention to certain information during the consultation process:
“The application dossier should contain sufficient information about the scientific validity and/or scientific rationale for the use of the biomarker, device measurement characteristics, device development characteristics, analytical and clinical performance.”
Source: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics
We have summarized information on the performance evaluation for you in a separate article.
Labeling
The CDx’s instructions for use must include the International Non-proprietary Name (INN) of the associated medicinal product that it is a companion test for.
Under German legislation, the CDx is not mentioned in the medicinal product’s patient information leaflet (AMG Section 11).
b) Specific features in the USA
The US authorities have more experience with CDx than the European authorities and notified bodies. The following specific regulatory features apply in the USA:
- The complete process is handled by the FDA and not by two authorities (notified body and EMA) as in Europe.
- Most CDx are “high-risk devices” and are classified as class III, which entails a requirement to submit a premarket application (PMA).
Only a few CDx can be authorized through a 510(k) application. - In an oncology context, the FDA also allows CDx to be intended for a specific group of medicinal products and not just for individual medicinal products. The requirement is that there is sufficient evidence that the IVD is suitable for this group of medicinal products.
There is an FDA guidance document available on this topic: Developing and Labeling In vitro Companion Diagnostic Devices for a Specific Group of Oncology Therapeutic Products.
- There is a List of Cleared and Approved Companion Diagnostics in the USA.
- Labeling: In the USA, the use of CDx together with a therapeutic product must be mentioned in the instructions for use and on the labeling of both the IVD AND the corresponding therapeutic product (see the FDA’s comments on this).
c) Guidance (or lack thereof)
There is still a lack of guidance on CDx. However, we recommend reading the following documents that are available already:
- Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics of the EMA
- Clinical Evidence Requirements for CE certification under the In-Vitro Diagnostic Regulation in the European Union from MedTech Europe
- MDCG 2020-16 rev. 2 Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 (in particular Annex II)
- MDCG 2022-10 Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
- EMA concept paper
- The EMA’s website provides information on implementation and further information on the role of EMA, including the application form and report template. However, it hasn’t been updated since February 2019.
- The pending sixth revision of the EMA’s Guideline on the clinical evaluation of anticancer medicinal products aims to provide better guidance on regulatory expectations for biomarker-driven development.
- FDA guidance on oncological research and CDx
- FDA recommendation on co-development: The development of biomarker-based diagnostic methods should be considered at an early stage of the clinical development of the medicinal product. The aim is to maximize the clinical applicability of the technology.
- MedTech Europe and the European Federation of Pharmaceutical Industries and Associations (EFPIA) have produced a document that lists the areas where the regulations for CDx are unclear and suggests solutions.
Given the lack of concrete European specifications, we recommend following the FDA’s specifications. The information from MedTech Europe on the subject of clinical evidence is helpful. In addition, manufacturers should always be aware of the latest developments. They can use the Johner Institute’s Regulatory Radar, for example, to make sure they stay up-to-date.
4. Bringing CDx to the European market
You can use the standard steps for authorization as a guide to safely launch a Companion Diagnostic on the market. However, note the specific requirements for CDx in each step.
a) Step 1: Define intended purpose as CDx
Use the intended purpose of your in vitro diagnostic device to check whether it is a CDx. This is only the case if there is an actual dependence on the therapeutic agent (see section 1 “Definition”).
b) Step 2: Quality management system
The next step is to set up your quality management system according to ISO 13485 and the IVDR. In this regard, there are NO differences from other types of IVD.
c) Step 3: Compile the technical documentation
There are also no specific requirements compared to other IVDs regarding the technical documentation according to Annexes II and III of the IVDR. The requirements of Annexes II and III of the IVDR must be met. See our comments on the performance evaluation (see the “Specific regulatory features” section).
IVD manufacturers should be in contact with their pharmaceutical partners during device development.
d) Step 4: Conformity assessment
The conformity assessment procedure is more complex than with other IVDs (see the “Consultation with medicinal products authorities” section). It starts with the formal application to the notified body. In addition to the name and classification of the CDx, the specific intended purpose including the reference to the specific medicinal product has to be specified, amongst others.
The drug authority, generally the EMA, is involved through the notified body. The EMA, the notified body, the IVD manufacturer and, if necessary, the pharmaceutical company should attend a pre-submission meeting intended to support the preparation of the application.
The consultation process follows the procedure described above (section 3a).
5. Tips for CDx development and approval
To ensure that everything runs smoothly during the development and approval of your Companion Diagnostic, we recommend the following:
a) Choose your partner well
Be discerning when choosing your partner from the IVD or pharmaceutical sector. Developing a CDx requires a huge amount of coordination and cooperation. For it to go smoothly, the framework conditions must be right. The CDx project will be easier if your partner has prior experience with Companion Diagnostics.
b) Reach clear agreements
Functioning agreements and a high level of communication are important for guaranteeing the compatibility of the IVD and therapeutic agent.
The planning should be coordinated to ensure that IVD development is consistent with the requirements concerning the medicinal product.
Prototype development, including the analytical performance evaluation, should be coordinated to ensure that the clinical trial can be started on time and as planned.
The EMA recommends that:
“The Agency recommends early interactions with the relevant notified body, the device manufacturer, and the marketing authorisation holder(s) or applicant(s) of the medicinal product(s) (as applicable and relevant).”
Source: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics
The FDA recommends that:
“Although codevelopment as a process does not require simultaneous development of the IVD companion diagnostic and the therapeutic product from beginning to end, the availability of an IVD with “market-ready” analytical performance characteristics (i.e., a test that is completely specified with complete analytical validation and meets the therapeutic product sponsor’s expectations for performance) is highly recommended at the time of initiation of clinical trial(s) intended to support approval of the therapeutic product.”
Source: https://www.fda.gov/media/99030/download
c) Involve the CDx in medicinal product development from an early stage
The alignment of the drug and CDx should be started as early as possible. It is therefore important for the medicinal product manufacturer to involve the IVD manufacturer as early as possible.
d) Plan more time than usual
If a Companion Diagnostic accompanies a medicinal product, both development and approval are more costly and complex than for a medicinal product or IVD on its own. The involvement of the drug authority also adds time to the procedure. These factors should be considered in your planning.
6. Conclusion
Companion Diagnostics (CDx) are becoming increasingly important in modern medicine. They play an importantrole in personalized medicine.
However, for manufacturers of IVDs or medicinal products, this means more efforts as CDx come with additional regulatory requirements. Therefore, good planning and effective coordination between all parties involved is crucial. But if their development is successful, CDx can bring huge benefits and take therapy to a new level for patients.
The Johner Institute’s experts will be happy to help you with any questions you may have on IVD authorization strategies or on the issue of Companion Diagnostics (CDx) specifically. Simply contact us via our contact form or email us.
Change history
- 2023-12-15: Chapter 3 c) addition of MDCG 2020-16 rev. 2 and MDCG 2022-10
- 2023-02-28: Chapter 1 b) Further information supplemented with link to MDCG 2020-16 rev.2
- 2022-01-07: Guidance on the procedural aspects for the consultation to the European Medicines Agency by a notified body on companion diagnostics of the EMA added
