It is not easy to get medical devices approved in Brazil. This is due to the number of regulations, their complexity, and the fact that Brazil has published most of the regulations in Portuguese only.
This article provides an overview and highlights similarities with the European and US systems. This will make it easier for you to understand and comply with the regulations in Brazil. Unnecessary rejections by the Brazilian authority and the resulting delay in approval can thus be avoided, making approval in Brazil faster and easier.
1. The Brazilian regulatory system for medical devices
ANVISA is the Brazilian agency responsible for medical devices. Comparable to the FDA,
- it issues laws on medical devices,
- reviews their approval, and
- monitors the conformity of the manufacturers’ QM systems with Brazilian requirements.
ANVISA’s requirements are similar to the European Medical Device Regulation (EU) 2017/745 and the US Quality System Regulations (21 CFR part 820):
- Manufacturers with a QM system according to 21 CFR Part 820 or ISO 13485:2016 mostly comply with Brazilian QMS requirements.
- Just as the EU and the FDA require registered local “Authorized Representatives,” a Brazilian Registration Holder is required.
- The agency divides medical devices into classes according to rules that are similar to those in Europe. These classes determine the effort and duration required for device approval. You can find out more about the classification process below.
In addition, Brazil has country-specific requirements, such as the requirement for an INMETRO certificate. More on this later.
2. Medical device legislation in Brazil
The following is an overview of the most important Brazilian requirements:
| Product type | Resolution | Content |
| Medical Devices | RDC 751/2022 | Registration, modification, maintenance, and withdrawal of medical devices |
| Medical Devices | RDC 270/2019 RDC 423/2020 | Requirements for registration of class I and class II devices |
| In vitro Diagnostics | RDC 830/2023 | Classification rules and requirements for registration and labeling of IVD |
| All | RDC 665/2022 | Brazilian Good Manufacturing Practices |
| All | Decree No. 8077/2013 | Regulates the approval and monitoring of devices by the authority in Brazil |
| All | Law No. 6360/1976 | General law on health surveillance |
3. Classification according to RDC 751/2022
Manufacturers intending to market a medical device in Brazil must first determine the risk class of the device. ANVISA distinguishes classes I (low risk) to IV (high risk).
The classification rules have been revised and are included in Annex I of Resolution RDC 751/2022. New rules address advanced technologies, including software as a medical device and medical devices made of nanomaterials. The rules are primarily adopted from the European MDR.
Use the following table as a guideline for classifying your device:
| EU | Brazil |
| I | I |
| IIa | II |
| IIb | III |
| III | IV |
The procedures for classifying and registering in vitro diagnostic medical devices (IVD) are not covered by this resolution and are part of RDC 830/2023.
4. Requirements for the approval of medical devices in Brazil
Before manufacturers submit the approval documents to the Brazilian authority, they must comply with specific requirements.
1. Requirement: The B-GMP certificate
Manufacturers must comply with the Brazilian Good Manufacturing Practices (B-GMP) requirements. These are similar to the FDA’s Quality System Regulations (21 CFR 820).
Manufacturers of class I and II devices must comply with these requirements but are not audited by ANVISA. They are also not required to submit a B-GMP certificate for device approval. By contrast, class III and IV device manufacturers must submit a B-GMP certificate for device approval.
Please note: Such a certificate can only be issued after an inspection, e.g., by ANVISA. Waiting for an audit from ANVISA can take several months.
With Resolution RDC-183/2017, adopted in October 2017, ANVISA has created significant simplifications for obtaining the B-GMP certificate:
- ANVISA recognizes MDSAP audits if the Brazilian requirements have been taken into account.
- ANVISA recognizes audit reports from recognized bodies from IMDRF member countries.
Submitting an audit report as described above can speed up the issuance of the B-GMP certificate and obtaining approval for your medical device in Brazil.
B-GMP certificates are valid for two years and must be renewed by the Brazilian Registration Holder (BRH) at least six months before they expire.
The costs of a GMP certificate account for a large proportion of the registration costs and can be around EUR 25,000 per period.
2. Requirement: The Brazilian Registration Holder (BRH)
An additional requirement for approval in Brazil is a local regional representative, the Brazilian Registration Holder (BRH). The BRH must be officially appointed and is legally responsible for the devices approved in Brazil.
A registered office in Brazil or a distributor can perform this task. Independent companies may also perform the tasks of the BRH.
Choose the BRH carefully! He will hold your registration! You depend on him.
The tasks of the Brazilian Registration Holder include:
- Communication with ANVISA
- Application for the B-GMP certificate
- Submission of marketing authorization documents
- Market surveillance (at least the BRH bears the responsibility for it)
- Vigilance: The BRH reports recalls and incidents to ANVISA
3. Requirement: The INMETRO certificate
INMETRO is the National Institute of Metrology, Quality and Technology. Some medical devices require a certificate for approval by ANVISA, which proves that a device complies with the applicable Brazilian standards and requirements.
INMETRO or an INMETRO-recognized body can perform the inspection and issue the associated certificate. The INMETRO certificate is valid indefinitely but must be maintained through regular audits.
Medical devices that require an INMETRO certificate include:
- Electrical devices
- Sterile injection syringes for single-use
- Breast implants
- Surgical and non-surgical gloves made of rubber
- Condoms
Allow three to 12 months to obtain your INMETRO certificate. Please also note that there are ongoing costs for the continuity of the certificate.
4. Requirement: The ANATEL certificate
ANATEL is the Brazilian national telecommunications authority. All medical devices with a radio module require an ANATEL certificate to be approved by ANVISA. Just like the INMETRO certificate, the requirement to submit an ANATEL certificate is not dependent on the device class. You also need an ANATEL certificate for a class I device if it has a radio module.
Currently, only Brazilian product tests are recognized by ANATEL. This means that you may have to have your device tested again for the Brazilian market.
Please refer to the detailed article on the INMETRO and ANATEL certificates. It also reveals how long these processes take, what they cost, and what mistakes manufacturers should avoid that could lead to unnecessary delays and difficulties.
5. Approval process
ANVISA distinguishes between two approval paths. These are based on the classification.
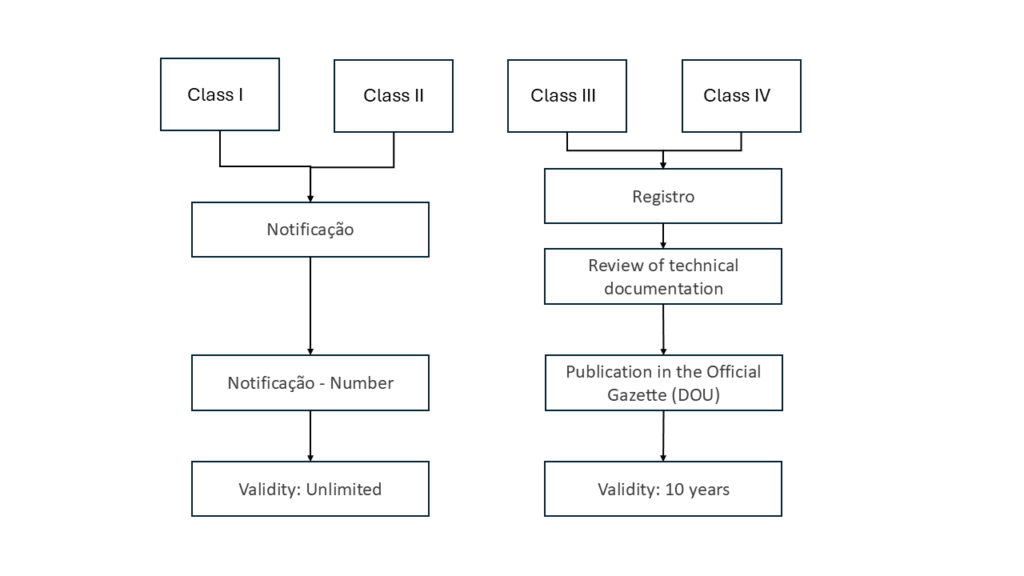
- Class I and II devices are notified to ANVISA and receive a registration number (Notificacao).
- Class III and IV devices undergo a complete approval and testing procedure (Registro).
6. Documentation requirements
The class and, thus, the approval procedures also determine the documentation requirements. The higher your device class, the more extensive the documentation requirements.
a) Requirements that apply to all classes
Regardless of the device class, manufacturers must comply with the (documentation) requirements of RDC 751/2022. These are based on the IMDRF’s ToC format and are summarized in Annex II of RDC 751/2022. Among other things, Annex II requires:
- A detailed description of the medical device, including the basics of how it works and how it functions, its contents or composition
- Indication, intended purpose, or application for which the medical device is designed according to the manufacturer
- Precautions, restrictions, warnings, special precautions, and explanations for the use of the medical device
- Information on storage and packaging
- Labeling samples and instructions for use
- Description of the manufacturing process
- Proof of compliance with the general safety and performance requirements
b) Requirements for the approval of medical devices in classes I and II
A simplified procedure is used to authorize class I and class II devices. In addition to providing general information about your device, you submit an assignment for the BRH and a confirmation of compliance with the B-GMP requirements. The authority issues a registration number without reviewing the technical documentation.
ANVISA is, however, permitted to request further documents.
Please note: If ANVISA conducts a review, you must provide the technical documentation to your BRH.
An unlimited authorization applies to class I and class II devices.
c) Requirements for the approval of medical devices in classes III and IV
ANVISA conducts a detailed review of the documentation for class III and IV medical devices. The authority requires manufacturers to submit large parts of their technical documentation. This includes, among other things:
- Device description
- Device specifications
- Information on risk management (risk management file)
- Software or firmware description
- Clinical evaluation
- Tests for biocompatibility, electrical safety, or electromagnetic compatibility (as applicable)
- Proof of usability
ANVISA will review the submitted documentation and, upon approval, publish it in the DOU (Diário Oficial da União). You will not receive an additional certificate.
The approval of class III and class IV devices is valid for 10 years.
If your GMP certificate loses its validity, the device registration will also become invalid! You must renew the GMP certificate every two years.
Chapter VII of RDC 751/2022 now defines the content requirements for creating the technical documentation. ANVISA has adopted the Table of Contents Format of the IMDRF for the specifications on the structure of the technical documentation in Annex II of the resolution.
d) Requirements for the approval of special medical devices in Brazil
For some devices, an economic report must also be submitted. These include orthopedic and cardiovascular devices.
The complete list can be found in decision RE n° 3385/2006 and on the ANVISA website. A helpful template for your economic report is also available on the website.
The economic report contains basic and economically relevant device information such as pricing, sales figures, and the estimated number of patients treated.
7. Conclusion and summary
The requirements for approval in Brazil are just as extensive as those in Europe or the USA. Companies can reuse most of the documents but must translate them into Portuguese. In addition, there are some requirements that are specific to Brazil.
As in Europe and the USA, the medical device class determines the duration and effort required for approval.
| Class I | Class II | Class III | Class IV | |
| Complexity | low | low | high | high |
| Duration | 1 month | 1 month | ~ 12 months | ~ 12 months |
| Validity of admission | unlimited | unlimited | 10 years | 10 years |
| B-GMP certificate | not necessary | not necessary | yes (valid for two years) | yes (valid for two years) |
The Johner Institute team can support you with the approval of your medical devices in Brazil. We will happily answer your questions, even free of charge, as part of our micro-consulting service.
Change history:
- 2024-10-15: Comprehensive revision incorporating new requirements from RDC 751/2022 and RDC 830/2023
- 2023-01-24: ANVISA issues RDC 751/2022, which establishes new rules for classifying medical devices and combines the rules for reporting, registering, modifying, renewing, and deleting registrations in a single resolution. It thus replaces various sets of rules, including RDC 185/2001.
- 2021-03-21: Note added to article about ANVISA and INMETRO certificates
- 2020-10-06: ANVISA eliminates the Cadastro route for class II devices with RDC-423/2020: Adaptation of Fig. 1 to the new system and update of the approval procedure in chapters 5 and 6.b)
