This FAQ provides answers to the most common questions that companies such as medical device manufacturers have about quality management systems (QM systems) and ISO 13485.
Also see the overview article on QM systems and ISO 13485, which links to other articles on the various activities involved in quality management.
Are you missing the answer to a question? Then feel free to benefit from our micro-consulting service.
Question 1: Who needs a QM system?
The EU Medical Device Regulations (MDR and IVDR) as well as the FDA set requirements for QM systems. These requirements are relevant for:
- medical device manufacturers
- distributors and importers who have manufacturer obligations under Article 16 of the MDR/IVDR
- organizations that reprocess or sterilize medical devices as new

The MDR and IVDR formulate these requirements in Article 10 (Section 9) and Annex IX.
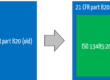
Until now, the FDA specified its requirements for QM systems in 21 CFR part 820.
With the new Quality Management System Regulation (QMSR), the FDA is pursuing harmonizing with the ISO 13485:2016 requirements.
The QMSR is now final and will apply from 02.02.2026. This means that manufacturers have two years to implement the requirements.
The transitional period for implementing the requirements of the new QMSR ends on 02.02.2026. Our expert team will happily help you with all the necessary preparations.
Question 2: Does the QM system have to be in conformity with ISO 13485?
For the European Union the short answer is yes, because EN ISO 13485 is the only QM standard that is harmonized for the MDR and IVDR.
With the FDA QMSR, the QM system must now also comply with ISO 13485:2016 and some additional FDA requirements for the US market.
Please also see our article on the FDA’s planned changes.
Question 3: Is ISO 9001 allowed as an alternative?
Other standards are permitted, but they are not suitable on their own for proving that the legal requirements for the QM system are met. ISO 9001 is, therefore, a standard that medical device manufacturers tend to fulfill in addition.
Other management standards, such as ISO 27001, do not focus on quality but, in this example, on IT security. These standards can or must also be fulfilled.
In such cases, ensure that you do not establish a parallel management system but an (!) integrated one. Benefit from our help.
Question 4: Does the QMS have to be ISO 13485 certified?
In the European Union manufacturers must involve a notified body in the conformity assessment unless the devices fall into class I/class A.
The most used conformity assessment route follows Annex IX of the MDR or IVDR. For this purpose, the manufacturers require an EU quality management system certificate.
Strictly speaking, no ISO 13485 certificate is required. But as ISO 13485 almost completely covers the QM requirements of Article 10 and Annex IX of the MDR and IVDR, it makes no sense to dispense with the ISO 13485 certificate.
Thus, the short statement that an ISO 13485 certificate is mandatory for all manufacturers (except those of class I/class A devices) for the European market is sufficiently correct.
For the FDA, conformity with ISO 13485:2016 and additional QMSR requirements is needed from February 02, 2026 but a certificate is not mandatory.
Manufacturers of devices in classes IIa and higher usually use the conformity assessment procedure in accordance with Annex IX. This requires a certified QM system. This is not necessarily the case with other conformity assessment procedures. However, these are only suitable in special constellations.
Contact us for quick help selecting the right conformity assessment procedure.
Question 5: What are the requirements of ISO 13485?
ISO 13485 obligates manufacturers to, among other things:
- QM manual, including quality objectives and quality policy
- Quality Management Representative (QMR)
- Description of standard documents such as standard operating procedures. These include, among other things, standard operating procedures on/for
- document control, including document release and retention periods
- management review
- internal audits
- corrective action and preventive action
- handling of measuring equipment
- management of suppliers, e.g., with quality assurance agreements (QAA)
- Work in conformity with these standard operating procedures
The requirement documents can be divided, for example, into Standard Operating Procedures (SOPs) and work instructions, templates, forms, and checklists. ISO 13485 requires approximately 25 SOPs to be documented, which you will find listed here.
Question 6: How to get the certification?
Step 1: Select certifier (notified body)
The German accreditation body DAkkS publishes on its website the notified bodies or certifiers accredited for ISO 13485 certification. Note that the notified bodies may only perform ISO 13485 certification for a certain type of medical device (e.g., in vitro diagnostic devices, non-active medical devices, extra-corporeal circuits).
Step 2: Establish quality management system
As described above, this includes creating the QM manual, describing standard operating procedures, and establishing the QM representative.
This page describes how you can establish a lean QM system in the shortest possible time with our help (scroll to the “7 steps”).
Step 3: Live quality management system
In order to apply for an audit, you must not only have defined a QM system, but you must already be working according to the rules of this (new) QM system. For example, technical documentation for a medical device should already have been developed following the respective SOPs such as the development SOP (as far as possible). Typically, you should also have performed an internal audit as well as a management review.
Step 4: Invite notified bodies for audit
You then invite the selected notified body to an audit. If you pass this audit, the notified body will (in all likelihood) issue you with an ISO 13485 certificate and, if applicable, an Annex XI certificate. Among other things, this authorizes you to market medical devices.
On this page, we have described how the path from your first contact to an ISO 13485-certified system can proceed.
Question 7: How long does it take to get the certificate?
We typically guide companies to ISO 13485 certification in six to nine months (more on the process). Currently, the overload of notified bodies may delay this timeframe. What matters most is the commitment of the organization.
Question 8: How much does it cost, what are the efforts?
The amount of time and effort required depends mainly on the size of the organization and the areas to be certified. Small and medium-sized companies spend between 30 and 90 days to set up their QM system.
With the help of the Johner Institute (Medical Device University & templates and/or consulting), companies succeed in significantly reducing this duration.
Maintaining the QM system on a permanent basis also means an expenditure, but this should be more than compensated for by lower consequential failure costs (recalls, rework, unnecessary iterations in development).
External certification costs are incurred in any case for the certification of their quality management system. For the preparation, the on-site audit, and the follow-up of the audit, the certification body will charge you costs that depend on the size of the company. These range from EUR 20 to 60,000. This includes the inspection of the technical documentation.
If you would use our support, there will be additional costs for this external consulting, which will vary depending on the size of the company, the current status of your quality management system, and the extent of your own work. We will be happy to provide you with a tailor-made offer!
Experience shows that our support also pays off financially because the costs for rework and interaction with the notified bodies are minimized.
Question 9: Where can I get support?
Johner Institute’s QM experts support medical device manufacturers and their service providers in setting up and continuously improving QM systems.
a) Education and training
The Johner Institute offers seminars for beginners and advanced users, e.g., on the basics of ISO 13485 or internal auditor.
b) Tool to master all regulatory hurdles
The Medical Device University enables manufacturers to create QM systems themselves with support. It combines:
- an onboarding that provides an overview of the roadmap
- video trainings
- complete set of sample templates that only need to be adapted to your own situation
- weekly expert sessions where questions are answered
c) Consulting and assumption of roles
Johner Institute consultants also provide direct support. They…
- create QM systems for and with you,
- check these systems and prepare them for audits (e.g., with mock audits),
- operate a QM system for manufacturers and act as legal manufacturer, and
- take on the role of QM representative(s).
Interested? Then contact us right away.
Change history
- February 2024: References to the FDA’s QMSR added
- August 2023: Answers to questions 1-3 slightly more detailed, editorial changes
- May 2023: First version created