The process FMEA (pFMEA) is a method for the systematic analysis of risks resulting from failure modes in processes, such as device production and cleaning.
Laws, such as the MDR, and standards, such as ISO 13485, require medical device manufacturers to identify and control such process risks.
1. What the pFMEA is
a) The pFMEA is a method for finding the effects of process failure modes
Like the design analysis (dFMEA), the process FMEA (pFMEA) is a variant of the FMEA (Failure Mode and Effect Analysis). Therefore, it is a bottom-up processthat analyzes the unknown effects of failure modes.
| process | process failure (example) | negative effect on the process outcome (example) |
| steam sterilization | temperature is not high enough | object is not sterile |
| printed circuit board assembly | wrong electronic component tape inserted | printed circuit board is incorrectly assembled, electronics do not function as intended |
| post-market surveillance | Person Responsible for Regulatory Compliance or bot does not find information source | report, e.g., PSUR is incomplete |
| static code analysis | analysis tool configuration file is overwritten | test report does not warn that the cyclomatic complexity of the code is too high |
b) The pFMEA is (generally) not a method for risk analysis
In the context of medical devices, the unknown and mostly undesirable effects of these failure modes are hazards, hazardous situations, and harm. If you know the probability and severity of the resulting harm, you also know the risks.
That’s why the FMEA is also generally understood as a method for risk analysis. However, this perception is not entirely accurate, as the method is only partially suitable for determining the severity of harm and its probability.
Furthermore, the effects are not harms as defined by ISO 14971 (e.g., physical injury to patients). Rather, they are elements in a causal chain that only later results in harm
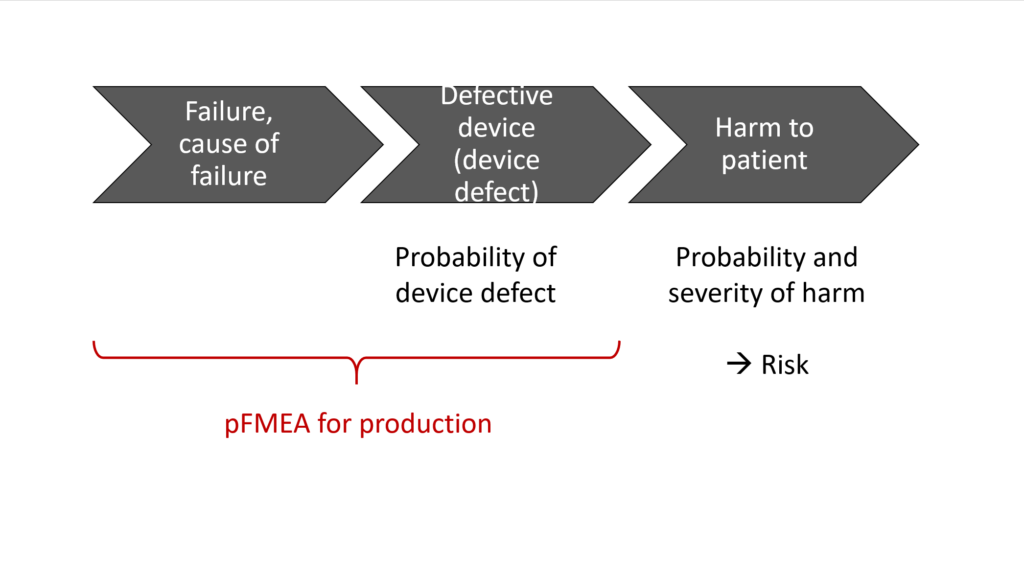
For example (see Tab. 1), a non-sterile device is not a case of harm in itself but rather a potential source of harm and, therefore, by definition a hazard and not a risk.
c) The pFMEA is a good complement to the dFMEA
As the acronyms themselves suggest, the pFMEA looks at the effects of deficient processes. In contrast, the dFMEA focuses on the effects of deficient products. The “d” in dFMEA stands for “design” in the sense of designing and assembling products, rather than in the sense of graphic design.
The result of a pFMEA is an estimate of the impact of a process failure on the outcome of the process. This (undesirable) process outcome could, for example, be a component that does not meet the specifications. This out-of-specification component would be the starting point of a dFMEA, as the dFMEA investigates the effects of out-of-specification components on the product as a whole and ultimately on patients.
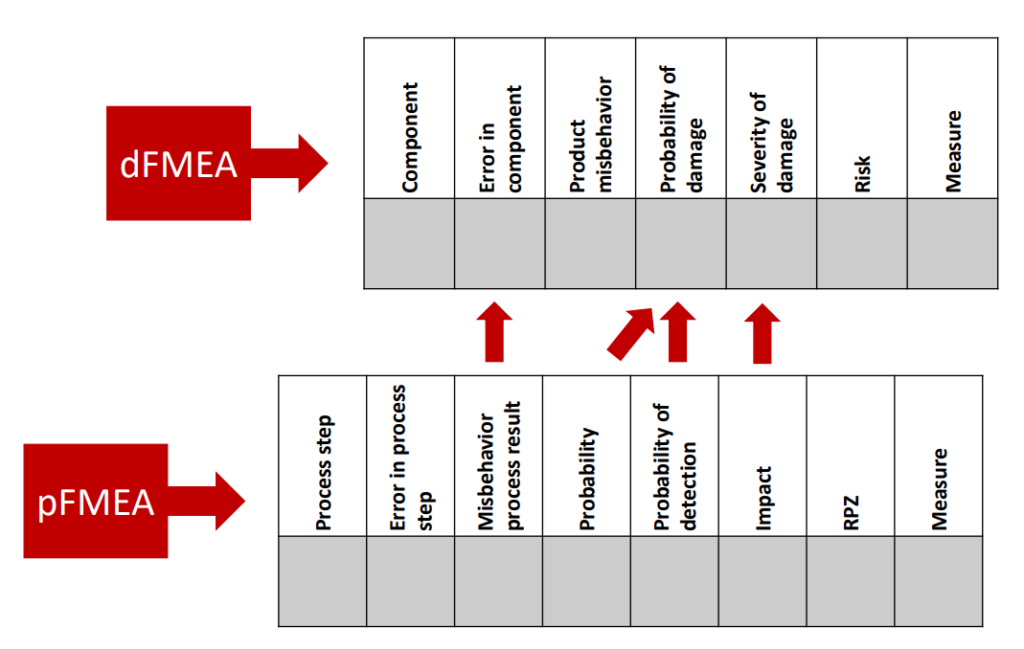
This means the pFMEA can act as the input for the dFMEA by providing essential information:
- Type of out-of-specification behavior (e.g., incorrectly assembled circuit boards)
- Extent of out-of-specification behavior (e.g., number of germs still on a sterilized part of the medical device)
- Probability of out-of-specification behavior
2. When the pFMEA should be used
a) pFMEA when designing new processes
Not just for core processes
Organizations should always perform a pFMEA when they are establishing new processes. This doesn’t just apply to core processes, such as the development, production, and maintenance of medical devices, but often to support processes as well, such as:
- Purchasing
- Supply, logistics, storage
- Monitoring of new and amended regulations
- Post-market surveillance
- Sales
- Education, training of employees and customers
If necessary, more than one pFMEA per process
Even for core processes, organizations shouldn’t limit themselves to one single pFMEA. Core processes can often involve several sub-processes or procedures that should be analyzed individually, as the following examples show:
- Development
- Modeling and simulation of physical properties, e.g., mechanical strength
- Software testing and static code analysis
- Collecting of training data and training of models in machine learning (artificial intelligence)
- Production
- Identification and traceability of components and materials
- Printed circuit board assembly
- Assembly of components
- Sterilization of devices
- Packing and labeling of devices
pFMEA for process design
The pFMEA aims to analyze the undesirable and unknown effects of deficient processes. Therefore, it can also help you to work out any necessary countermeasures, for example additional test steps. And these, in turn, can lead to changes in the processes, meaning that the process design, pFMEA and process change are iterative.
b) pFMEA when changing existing processes
There are a lot of occasions when processes have to be changed:
- Previously unknown risks have come to light
- Known risks were assessed incorrectly (too low)
- Process inputs change, e.g., because components are no longer available
- Process elements have to be changed, e.g., a software update in production line control electronics
- The process outputs have to be changed
- Manufacturers have to increase process automation for economic reasons
All these changes have to be evaluated by the organizations to see if they could result in
- new (unknown) effects,
- known effects with a different probability, or
- a change in severity.
This is exactly what the pFMEA is intended for.
c) pFMEA for the modification of existing processes
The pFMEA is more than just a necessary consequence of a process change. It can also be the trigger for a change. This is because an organization can use the pFMEA to identify weak points in their processes and how to improve them, even if there are no negative effects on patients, users, and third parties.
Improvements could relate to, for example:
- The number of non-critical errors
- The need for rework and testing steps
- The duration of the process
- Energy and material use
- Differentiation from and interaction with other processes
- Deciding for or against outsourcing
d) pFMEA when preparing the process validation
The regulatory requirements (see chapter 4 of this article) require manufacturers validate their processes. This validation often means high costs for manufacturers. And complete testing of all process parameters or the complete testing of process software is generally impossible in reality.
But legal requirements and standards allow for a risk-based approach. In other words, the manufacturers are allowed to adjust the time and cost required for the validation of the process in line with the risks generated by the process.
The pFMEA helps to quantify these risks, even if it is not a risk analysis method as defined by ISO 14971.
In this article on process validation it is explained in detail as well as which regulatory requirements have to be met.
3. How the pFMEA works
Step 1: Planning
When planning the pFMEA, the organization has to define the following:
- The process to be analyzed
- The team that will perform or be involved in the pFMEA
- The activities required (these are the ones described below)
- Time and duration of the pFMEA
- Regulatory requirements that must be met
- If applicable, methods and tools for, e.g., documentation
Step 2: Preparation
To prepare for the pFMEA, the team first has to review and update the process description. If this description hasn’t been created, the team has to create this documentation.
The process description should include:
- Process steps. For each step:
- The inputs (materials, energies, information)
- The outputs (materials, energies, information)
- The people involved
- The planned activities with associated work instructions and requirements
- The systems involved (tools, machines, software, etc.) with existing validation records, if applicable
- Process outcome requirements
- For each process outcome, an assessment of the consequences of it not being met. To keep things simple, each process outcome can be classified as critical or non-critical. The classification can be based, e.g., on a dFMEA.
Diagrams in, for example, Business Process Modeling Notation (BPMN) are a good way of clearly documenting these processes.
Step 3: Execution
The actual pFMEA involves looking at each process step and working out which failure modes can occur and what consequences they would have for the process outcome. The following questions can be used to guide the process:
- What are the consequences for the output of a work step if
- their inputs do not comply with the specification?
- their activities are not carried out or not carried out as specified?
- a system (machine, tools, etc.) does not behave as specified?
- What are the consequences if a work step is not performed, or two work steps are performed in the wrong order?
- How can you tell if one of the above failure modes has occurred?
The Ishikawa method will help you identify possible sources of failure modes in each process step. The HAZOP method (IEC 61882) is very useful for describing the different forms of deficient inputs and outputs.
The results of the pFMEA must be documented. This can be done in a table:
| Process step | Process step failure mode | Cause of failure, if applicable | Probability of failure | 1 – Probability of failure being detected | Effect of the failure | RPN | Actions |
| 1: … | |||||||
| 2: … |
The three following variables are quantified in the pFMEA (values between 0 and 10):
- Probability of the failure mode occurring
- One minus the probability of detection – if the probability of detection is 1 (i.e., 100%), the value in the column is zero.
- Effect of the failure mode (0 = no effect, 10 = effect of maximum severity)
The Risk Priority Number (RPN) is the product of the three quantitative variables and therefore has a value between 0 and 1000.
Many companies use 125 as a limit value below which no risk-mitigating measures are necessary. However, ISO 14971 and the MDR and IVDR insist that risks must be reduced as far as possible. Therefore, this number would at least have to be justified. It would be better to dispense with it and decide individually for each “risk” whether measures are necessary.
Step 4: Evaluation of the result
The RPN provides guidance on the extent of the failure mode’s impact. However, it is not considered a measure of risk, but rather a tool for prioritizing tasks (especially actions).
The final assessment of the risk in the sense of ISO 14971 is no longer (solely) the responsibility of the “pFMEA team.” Instead, the consequences of the deficient process outputs must be extrapolated to the patient, users, and third parties.
Step 5: Sharing of results
The main outcome of the pFMEA is the table shown above. However, the actions are no longer considered part of this analysis. The “action” column is nevertheless useful for ensuring that the actions for the risks identified have actually been implemented.
4. Which mistakes should you avoid when carrying out a pFMEA
Mistake 1: Unclear goals
The team should have a clear and shared understanding of the goals, including:
- The “scope,” in particular the process (i.e., production as a whole or “just” assembly?)
- The objective (e.g., is it to ensure compliance with regulatory requirements or to improve the process?)
- The importance of the pFMEA (is patient harm a possible consequence of an inadequate pFMEA or would it “just” result in a slow process?)
- The regulatory requirements that must be met by or during the process (e.g., FDA or EU requirements?)
Mistake 2: Team poorly assembled
The team should include the following roles, at least some of the time:
- Risk manager
- Is on top of the methodology
- Knows the effects of a suboptimal process output on patients, users, and third parties
- Knows the regulatory requirements
- Process owner
- knows the process
- People involved in the process
- know what can go wrong
- Quality manager
Mistake 3: Wrong point in time chosen
The pFMEA should be used:
- When the process is first designed
- If feedback from downstream phases gives reason to do so
- If the process, including the systems involved in it, is being changed
- Regularly, to confirm the “state of the art”
Mistake 4: Lack of coordination
The pFMEA alone will not generate the information needed to identify and quantify risks from medical devices. Rather, the following must be considered:
- All processes that provide inputs to the processes being reviewed (e.g., component production for assembly)
- All processes that work with the process outputs (e.g., production with results of development)
- Overall risk management that must include development (dFMEA) as well as production and downstream processes (pFMEA)
This often requires several process owners to coordinate, even if only one process is changed.
Mistake 5: Misunderstanding the RPN and its factors
The RPN is not a measure of risk according to ISO 14971. The “effect” factor should not be confused with the severity of harm according to ISO 14971.
The multiplication of three numerical values often creates a false illusion of accuracy. But, generally, each of the factors is based on a rough estimate.
5. The regulatory requirements the pFMEA helps you meet
Manufacturers must identify and manage the risks generated by their medical devices in line with the state of the art. The pFMEA is a state-of-the-art method.
These requirements are found in, for example:
- MDR, IVDR
- Article 10 (2)
- Annex I (3)
- Several other sections of Annex I, including (10) and (11)
- Annex II, Sections (3), (5)
- ISO 13485
- Requirements for a risk-based approach, e.g., when validating processes, measuring equipment, and computerized software
- Process validation specifications, including Section 7.5.6
- FDA 21 CFR part 820 (which will be replaced by ISO 13485)
Read more on computerized software validation, which can and should also be risk-based and for which a pFMEA is a valuable method.
6. Conclusion and summary
The pFMEA is used to systematically analyze the potential effects of a failure mode within a process.
Some auditors consider the pFMEA to be state of the art. Regardless of this, organizations should consider the pFMEA as a method, especially for processes that have an impact on the conformity of devices, e.g., production.
With pFMEA, organizations not only achieve compliance but can also structure their efforts, e.g., for product testing, on a risk-based basis, thereby avoiding unnecessary effort.
The Johner Institute helps medical device manufacturers and their service providers set up their QM systems and with the risk analysis for their devices and processes. Get in touch with us.
We also provide several videos and accompanying templates that manufacturers can use to perform legally compliant dFMEAs and pFMEAs in the Medical Device University.
Change history:
- 2025-05-14: Only minor editorial changes, mainly redesigned info boxes, links added, spelling mistakes corrected
- 2022-05-03: Article published in its current form