What is a Device Master Record (DMR)? Does software also need a DMR? If so, what are the regulatory requirements in the US and Europe? And what should a DMR contain?
This article provides answers.
With the introducing of the new Quality Management System Regulation (QMSR) in February 2026, the FDA will not any longer require the establishment of a Device Master Record. From then on, records according to ISO 13485, such as the “medical device file,” will be required.
What is a Device Master Record?
‘Device Master Record’ (DMR) is a term introduced by the FDA that is also used outside the United States.
A DMR is understood to be a “file” that, to put it simply,
- describes the device and
- explains how the device is to be manufactured, used, and maintained.
A device master record should, therefore, contain the following:
- Intended use
- Specifications (of the device and components), system architecture, illustrations, calculations
- Information on manufacturing: tools, methods, (inspection/acceptance) procedures, specifications, and requirements for production
- Procedures for quality control: specifications for how the device is inspected or tested (especially in production), e.g., with which methods and tools. For software, where there is effectively no production, these could be instructions for verifying the successul installation or configuration.
- Information on “labeling”. This includes labels, for example, on thepackaging, or on the device, instructions for use, further manuals, usage of symbols.
- Information on installation, maintenance, service, storage, and transport, including methods and tools
The results of the design output (e.g., specifications) also “end up” in the device master record.
The DMR should not be confused with the DHR, the Device History Record, which must be created for each product instance or batch.
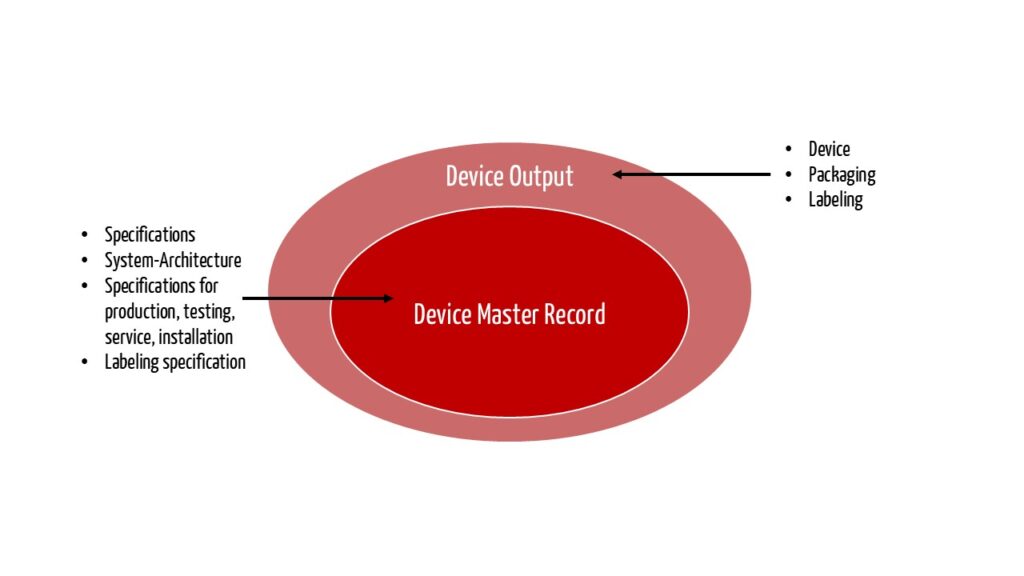
The FDA states the following about the relation between DMR and Design Output:
„The total finished design output consists of the device, its packaging and labeling, and the device master record.“
Which regulatory requirements apply to a DMR?
In the USA, the Device Master Record is mandatory. In Europe, there are comparable requirements in the form of the “Technical Documentation” and the “Medical Device File” of ISO 13485.
Requirements in the US
The FDA defines the Device Master Record as a “compilation of records containing the procedures and specifications for a finished device”. 21 CFR part 820.181 describes the required elements of a DMR. A summary has already been provided above.
Requirements of ISO 13485:2016
Compared to its previous version, ISO 13485:2016 introduces a Medical Device File (MDF) similar to the Device Master Record. It requires similar information to that described above.
In addition, the standard requires traceability of changes to the product over its lifetime. This means the MDF should always contain the currently valid product descriptions (according to the list above).
If you want to track the individual versions and changes in the DMR or MDF, the file must be updated with each product version. To have the required traceability, it must be possible to restore the state of the DMR for each older version.
Requirements of the EU Medical Device Regulation (MDR) 2017/745
The MDR places requirements on the technical documentation that contains elements of a DMR.
Do we need a DMR for software? If so, what should it contain?
For each medical device, you need a Device Master Record or its ISO 13485 counterpart, the Medical Device File. If the software is part of your product, then the software specification or requirements are part of the DMR.
For standalone software, the DMR includes the design output and further information.
Design output for software (as part of the DMR)
Further elements of a DMR for standalone software
- Intended use
- Instructions for use
- Installation instructions
- Instructions for production, e.g. production and deployment procedure incl. tasks such as
- storing the software on on media such as DVDsUploading to an app store
- description of any specific restrictions that affect certain jursdictions, operating systems, hardware
- Information on whether/how the software will be updated
- How long can a software version be sold on a DVD, for example? (DVDs age)
- How are updates distributed?
- How is their installation ensured?
- How are old versions withdrawn?
- Packaging specifications
- Training materials
- License information
Differentiation from the Design History File (DHF)
The Design History File would also include the following documents (including their versions)
- Specification and results of software tests such as unit tests, integration tests, and software system tests
- Results of static code analysis and code reviews
- Specifications, verification, and validation of usability engineering
- Software release
- Risk management file
- Instructions for creating the software, including build scripts and configuration files
The objective of the DHF is also to prove that the software has been developed in accordance with the development plan.
Do you need help compiling your DMR, MDF, or technical documentation? Then contact us right away.
Change history:
2024-11-18: Revision due to the introduction of the new QMSR

{kind=link}