The third edition of ISO 14971 has been available since December 2019.
This new version of ISO 14971 was published as ISO 14971:2019. It is an evolutionary development of ISO 14971:2007 and does not break with previous concepts.
Manufacturers should familiarize themselves with the new and amended requirements of this standard. In December 2019, the FDA recognized the third edition of the ISO 14971 standard. More on the transition periods below.
1. Third edition of ISO 14971
The third edition of ISO 14971 follows its predecessor ISO 14971:2007 (“second edition”).
At the same time, ISO has also revised ISO 24971, which is also available as a draft. This “explanatory standard” is becoming more important because it now contains some of the non-normative annexes of the old ISO 14971.
2. Overview of the changes
a) New chapter structure
The first thing that stands out is the new chapter structure. ISO 14971:2019 now follows the usual structure, which starts with the chapters:
- Scope
- Normative references
- Terms and definitions
The new chapter with the normative references changes the numbering: ISO 14971:2019 now has ten chapters.
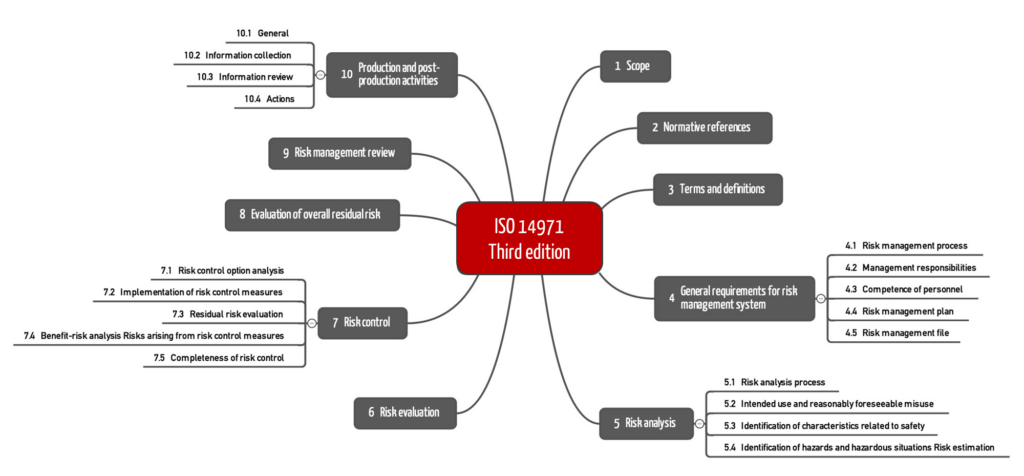
The chapter structure reveals another difference: The requirements for the post-production phase are more comprehensive and divided into four sections (10.1 to 10.4).
b) Higher relevance of the benefit-risk ratio?
ISO 14971:2019 claims to place even greater emphasis on demonstrating that the benefits outweigh the risks. It adds the missing definition of the term “benefit.”
“positive impact or desirable outcome of the use of a medical device on the health of an individual, or a positive impact on patient management or public health”
Source: ISO 14971, 3rd edition
Examples of these benefits are:
- Faster recovery, more complete recovery
- Curing with fewer side effects
- More accurate diagnosis
- Better public healthcare
This makes it clear that the benefit refers to a medical benefit and not, for example, a higher economic benefit for the operator.
The standard does not really establish any new requirements. It continues to state that it is the management’s job to determine the risk policy. It must be based on the state of the art. The third edition of ISO 14971:2019 at least adds a definition of the term “state of the art.”
“developed stage of technical capability at a given time as regards products, processes and services, based on the relevant consolidated findings of science, technology and experience”
Source: ISO 14971, 3rd edition
This state of the art cannot be compared with the state of the science. Instead, it is more in line with generally accepted technical and medical “good practices.”
One novelty of the third edition of ISO 14971 is that the manufacturers can define acceptance criteria for the evaluation of individual risks that are different to those used for the evaluation of the overall residual risk. The acceptance criteria for the individual risks can be used to decide on the need for risk control measures. The acceptance criteria for the overall risk can be used to decide whether the device can be marketed.
The standard obliges manufacturers to describe the methods used to determine the acceptability of the overall residual risk.
c) IT security in scope
The third edition of ISO 14971 explicitly includes risks resulting from inadequate “data and system security.” However, it does not define any specific requirements.
In German-speaking countries in particular, there is a risk that manufacturers won’t distinguish precisely between safety and security because both terms are translated as “Sicherheit” in German.
While weighing medical benefits against “safety risks” makes sense, weighing medical benefits against “security risks” can lead to confusion. An increase in security can even have negative effects on safety.
Don’t leave the safety of your patients to chance. Play it safe with a pentest from the Johner Institute!
d) Reasonably foreseeable misuse must be taken into account
ISO 14971:2019 adds the explicit requirement to analyze reasonably foreseeable misuse. It defines this “reasonably foreseeable misuse” as follows:
“use of a product or system in a way not intended by the manufacturer, but which can result from readily predictable human behavior”
Source: ISO 14971, 3rd edition
Such misuse can be intentional or unintentional. An example would be using a medical device without carefully reading the instructions for use before.
e) Safety-related characteristics must be identified
The chapter on safety-related characteristics may be new, but the requirements are not. Manufacturers must record these device characteristics in terms of quality and quantity – ideally with details of the limit values essential for the device’s safety. All IEC 60601-1 experts will immediately think of the essential performance characteristics. And rightly so.
The Johner Institute recommends that the system requirements, in particular, should be examined in order to determine whether there could be any risk if these requirements are not met or not met to the specified extent.
f) Production and post-production requirements
The most obvious change relates to risk management in production and the post-production phase, i.e., the post-market phase. The requirements are very similar to those of the MDR.
Both the MDR and the third edition of ISO 14971 require proactive collection and evaluation of data from post-development phases. The MDR talks about a process, ISO 14971 about a system.
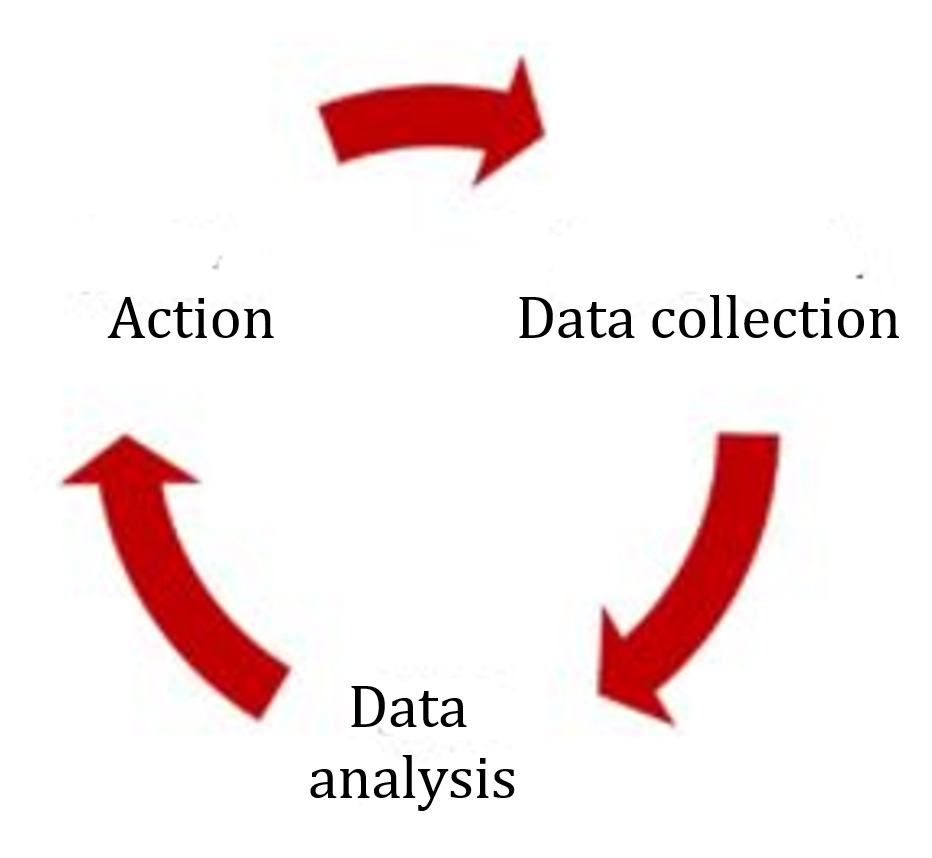
Similar to the MDR, the standard also defines the sources of information that always have to be considered, such as public information, information on the state of the art, and information generated during the installation, use, and maintenance of the device.
The information must be used to determine whether
- new hazards not previously considered have to be taken into account,
- the risks (probabilities and severity of damage) have been correctly assessed, and
- the risks are still acceptable, e.g., because the state of the art has changed.
The manufacturer must then act based on the results of this evaluation. The third edition of ISO 14971 mentions actions relating to the medical device (e.g., implementation of new risk-minimizing actions) and actions that relate to risk management (e.g., risk management process).
Read more on the subject of the post-production phase and post-market surveillance.
3. Legal obligation
a) Europe
EN ISO 14971:2019 has now been harmonized for both MDR and IVDR. The implementing decisions on harmonization can be found online for the MDR and IVDR.
b) USA/FDA
The FDA will continue to allow the second edition of the 2007 standard until the end of 2022. After that, at the latest, the FDA will insist on the application of the third edition of ISO 14971.
4. Conclusion
The third edition of ISO 14971 is even better than the already good second edition. Many of the changes are editorial, providing greater clarity and stringency.
Particularly noteworthy are the more precise requirements for the post-production phase. Nevertheless, the scope of the changes remains so limited that “version 2.1” would perhaps have been more appropriate. Particularly regrettable is:
- A lot of helpful annexes have been moved to ISO 24971. This does make ISO 14971 more lean, but forces manufacturers to buy a second standard.
- Some explanations have disappeared completely. The old ISO 14971 had made it clear that risk is not calculated by simply multiplying the severity and probability of damage. How can such a central and justified statement be taken out given that 95% of manufacturers do exactly that error?
- It can be assumed that the EU considers the requirements of the MDR regarding risk management to be only partially covered by the third edition. This means, there is once again the threat of additional requirements and normative interpretations in the Z-annexes.
- The interaction of risk management and the clinical evaluation is not described at all in the third edition of 14971, and only described in very basic terms in the revised ISO 24971.
- It is understandable that the standards committee wanted to align the standard with the usual chapter structure. This editorial change means that most manufacturers will have to check their specification documents (SOPs, work instructions, templates, etc.) for correct references to the chapter structure. A lot of work that does not contribute to patient safety.
Despite these downsides, manufacturers should be able to live well with this third edition of ISO 14971. Sometimes less really is more.
5. Latest news
The German version, DIN EN ISO 14971:2022-04, has also been published since spring 2022. This includes the harmonized version EN ISO 14971:2019 and Annex A11:2021, which primarily clarifies once again that the standard is compatible with the MDR and IVDR.
Change history:
- 2022-08-25: Reference to German version DIN EN ISO 14971:2022-04 and harmonization added
- 2020-11-16: Reference to Standardization Request and, thus, to the planning of harmonization of EN ISO 14971:2019 added