The EU Medical Device Regulation uses standalone software to describe “devices in the form of software.” However, this regulation only applies to some standalone software.
Content
This page provides a brief overview and references articles for further background information.
- Taxonomy
- Regulatory requirements for standalone software
- Five challenges and possible solutions
- Support
1. Taxonomy of software devices
a) Definition
Standalone software applications are independent devices brought to market without hardware, either as a download (e.g., via an app store) or on a physical data carrier (e.g., flash drive).
b) Delimitation
Standalone software intended for the healthcare sector is different from health software. It is also incongruent with medical device software (see Fig. 1).
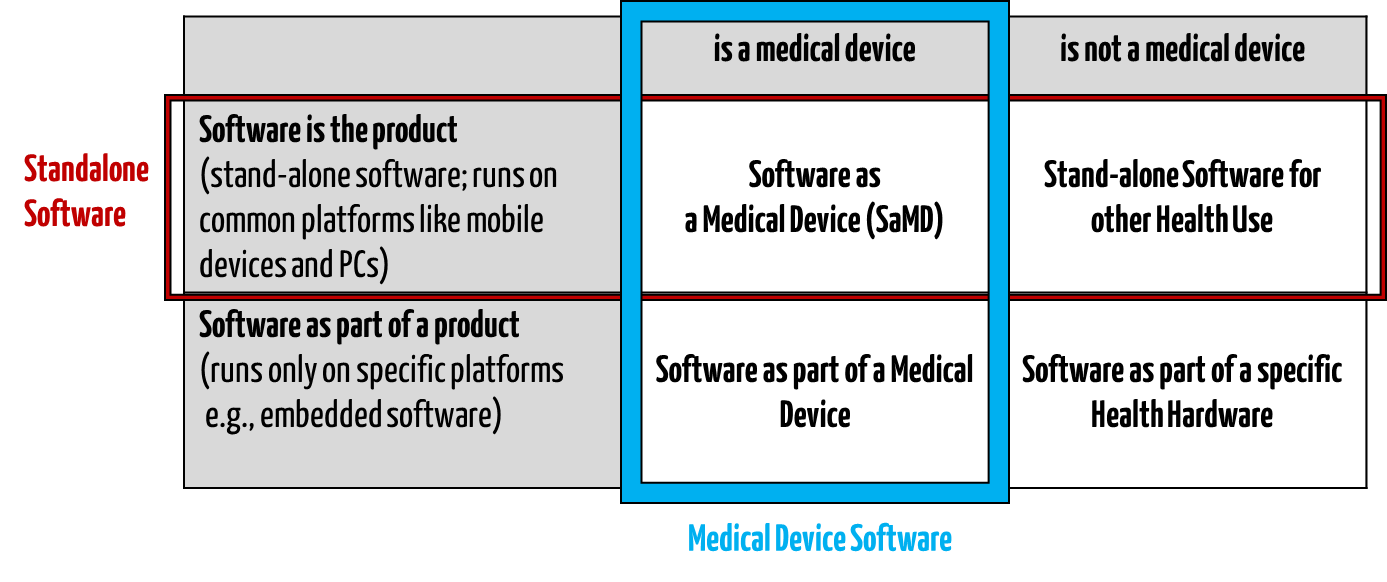
Fig. 1: Standalone healthcare software is a subset of health software (all four quadrants).
c) Examples
Examples of standalone software are
2. Regulatory requirements
a) Standalone software – a medical device?
Manufacturers must clarify whether their standalone software counts as a medical device. The article on the qualification and classification of software as a medical device sheds light on when this is the case.
b) Regulations, laws, standards
If the standalone software counts (“qualifies”) as a medical device , it must meet the legal and normative requirements. These do not differ from the requirements for software that is part of a medical device.
- The medical device regulations (MDR, IVDR) are relevant in Europe. These contain general regulations. This article presents the essential requirements for software.
- IEC 62304 defines the life cycle processes for medical device software.
- IEC 82304-1 applies to all health software and, therefore, all standalone software in the healthcare sector. It requires conformity with the requirements of IEC 62304.
- The FDA also sets out specific requirements for medical software in its guidance documents.
3. Five challenges and possible solutions
Challenge 1: Identification of the standalone software
With web-based medical devices in particular, manufacturers find it challenging to determine which part is part of the medical device and which is part of the runtime environment. For example, is the application server included?
It is important to document this definition explicitly.

Fig. 2: Web-based medical devices consist of many levels. One part belongs to the software (medical device), one part to its runtime environment. SOUP are part of the medical device.
Challenge 2: Placing on the market
If the software is made available via app stores, the question arises as to when the placing on the market actually occurs. When it is uploaded to the store? When it is activated in the app store? Or only at download?
This article on placing on the market provides answers.
Challenge 3: Software testing
The runtime environments on which the standalone software is installed differ. Hardly any two computer systems are the same. This applies to notebooks and servers as well as smartphones. There are thousands of Android-based end devices.
This makes it difficult for manufacturers to review the correct functioning of their software on these end devices. They therefore have to restrict them or test them riskbased.
Challenge 4: Actual use
This diversity affects not only the technical environment, but also the use environments and benefits. How are manufacturers supposed to ensure that only the users intended for the intended purpose use the devices? How should they anticipate under which circumstances (e.g., when driving, at night, during sport) their devices will be used?
Systematic post-market surveillance is essential here in order to track actual use and react to it if necessary.
Challenge 5: Latency of “approval”
In contrast to many physical devices, software must and can be brought to market in short development cycles. This is necessary because security patches must be installed.
However, this is offset by lengthy approval and conformity assessment procedures.
The Johner Institute digitizes the regulatory processes and works on real-time regulation.
4. Support for software manufacturers
Benefit from the support of the Johner Institute:
Contact us right away so that we can discuss the next steps. This will ensure that the “approval” process succeeds and that your software or devices are quickly launched on the market.