1. Definitions
Medical software includes all software used for healthcare, particularly for medical devices or medical devices (embedded software), and software that is itself a medical device (standalone software).
IEC/CD1 82304-1 (Health Software – Part 1: General requirements for product safety) distinguishes between the following terms:
- HEALTH SOFTWARE
Software intended to be used specifically for maintaining or improving health of individual persons, or the delivery of care
- MEDICAL SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device or intended to be a SOFTWARE MEDICAL DEVICE
- SOFTWARE MEDICAL DEVICE
Software intended to be a medical device in its own right
- MEDICAL DEVICE SOFTWARE
Software intended to be used specifically for incorporation into a physical medical device
This clarifies that medical software can be a medical device but does not have to be.
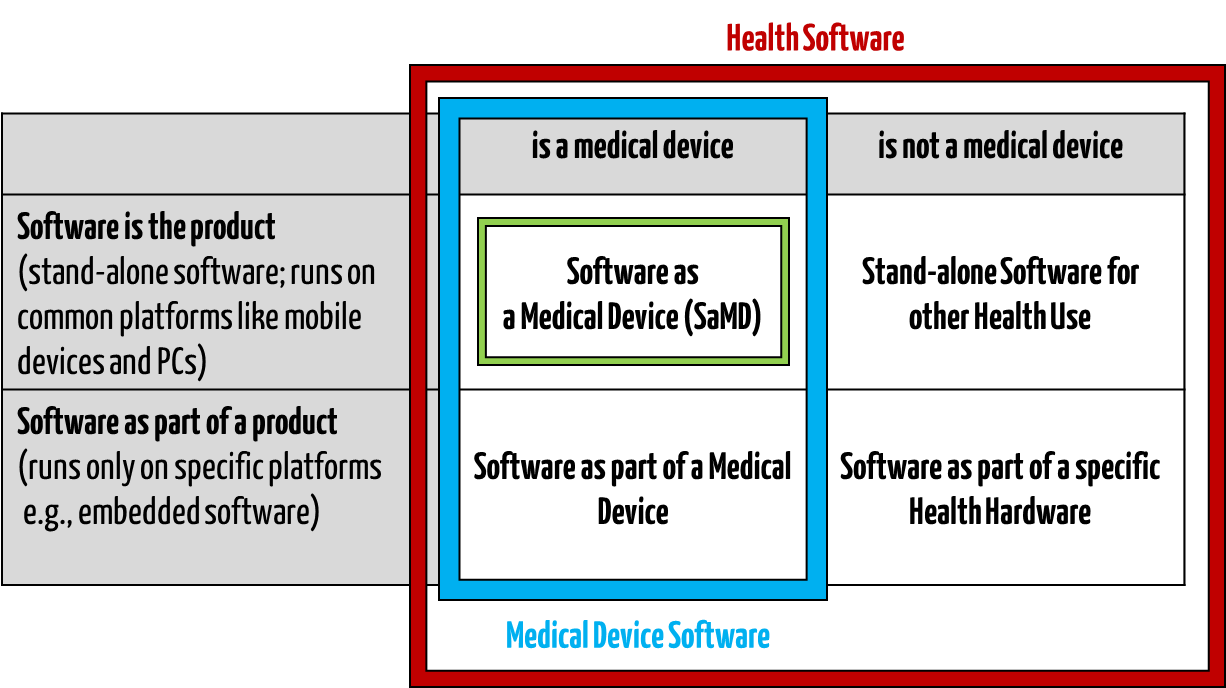
Fig. 1: Medical software includes medical device software and software as a medical device (click to enlarge).
2. Regulatory requirements
a) Medical software – a medical device?
The question often arises as to when medical software meets the definition of a medical device. You can find a further discussion on this topic in the article on the classification of software as a medical device and in the article on the qualification and classification of IVD medical device software.
b) Regulations, laws, standards
Software that is a medical device or part of a medical device must meet the regulatory requirements:
- In Europe, the medical device regulations (MDR, IVDR) are relevant. However, these only contain relatively general regulations for software, which this article presents.
- IEC 62304 defines the life cycle processes for medical device software.
- IEC 82304-1 applies to all “health software”. IEC 82304-1 also requires conformity with the requirements of IEC 62304.
- There are also MDCG guidelines, e.g., MDCG 2019-11 and MDCG 2023-4.
- The FDA sets out specific requirements in its guidance documents, including specific requirements for medical software. It also answered many questions specifically about software as a medical device in this FAQ.
3. Support for medical device manufacturers
Benefit from the support of the Johner Institute:
Contact us right away so that we can discuss the next steps. This will ensure that the “approval” is a success and that your software or devices are quickly launched on the market.
Accessibility refers to the design of products and services that can also be used by people with physical limitations. The term “products and services” encompasses both physical and digital products and services. This also includes medical devices (physical devices, apps, other standalone software). This article explains which accessibility requirements manufacturers of medical devices should be…
Details
Cyclomatic complexity is a metric in software engineering that helps determine the complexity and, thus, the likelyhood of errors and the maintainability of code. Cyclomatic complexity is also referred to as the McCabe metric.
Electronic and digital signatures should be considered on an equal level to handwritten signatures (“wet ink”). The requirements that need to be fulfilled depend on the extent of the binding force that is to be achieved and so depend on the document that is to be signed. This article explains
Details
PDMS stands for patient data management system. These clinical information systems are typically used in hospitals, especially in departments that treat patients in intensive care. PMDS are experiencing a new boom in Germany as a result of the funding provided by the Hospital Future Act (Krankenhaus-Zukunftsgesetz, KHZG). This article provides
Details
What is the difference between verification and validation, and how are these terms defined? Even standards and regulations use the terms incorrectly or misleadingly. This article
Details
Laws require risk management in hospitals, especially in order to improve patient safety. Nevertheless, many hospitals find this difficult. This article presents the most important regulatory requirements and provides tips for implementation.
Details
The qualification and classification of IVD software determine how and how quickly IVD manufacturers can bring their software to market and what costs arise for “approval.” This article will help you correctly qualify and classify IVD software, thereby avoiding regulatory problems and the resulting costs and delays.
Details
The Medical Device Regulation (MDR) (like the Medical Device Directive (MDD) and thus the Medical Device Act before it) requires manufacturers to comply with life cycle processes for their software. IEC 62304 and IEC 82304 also refer to software life cycle processes. But what is a software life cycle?
1. Documentation Level: End of Level of Concern On June 14, 2023, the FDA released the guidance document Content of Premarket Submissions for Device Software Functions. This document replaces the guidance document introducing the Level of Concern and only distinguishes between two classes.
Details
Software maintenance is the phase in which software is further developed, e.g., with the objective of According to the FDA, 79% of all bugs occur during software maintenance. Accordingly, some regulations address this topic.
Details