Before manufacturers place a medical device on the market in the EU, they must undergo a conformity assessment procedure (not quite correctly referred to as an approval procedure).
Content
This page provides a quick overview and contains links to relevant articles.
- Conformity assessment: The basics
- Course of the conformity assessment procedures
- Further articles on conformity assessment
- Support throughout the procedure
1. Conformity assessment: The basics
a) Objective and definition
The objective of conformity assessment is for medical device manufacturers to evaluate (themselves!) the conformity of their devices with the general safety and performance requirements of the EU medical device regulations (MDR and IVDR) and, if successful, to declare conformity – with a declaration of conformity.
Definition:
“conformity assessment’ means the process demonstrating whether the requirements of this Regulation relating to a device have been fulfilled;”
MDR
Manufacturers must involve a notified body in the conformity assessment except for class I devices.
b) Conformity assessment procedure
Depending on the class of the medical device, manufacturers can select a suitable conformity assessment procedure (see Fig. 1).
The following table provides an overview of these conformity assessment procedures:
| Description of the procedure |
MDR, IVDR |
| Manufacturer sets up a complete QM system and has it certified according to ISO 13485 and the Annex (see right) |
Annex IX |
| Manufacturer prepares technical documentation and declares conformity |
Annex IV |
| Manufacturer has prototype tested by notified body |
Annex X |
| Manufacturer sets up a QM system for production |
Annex XI part A |
| Manufacturer has every device produced tested by notified body |
Annex XI part B |
| Manufacturer establishes quality assurance (final inspection) |
— |
Compared to the now obsolete EU MDD Directive, the EU MDR and IVDR regulations no longer have a conformity assessment procedure for production based solely on the final testing of devices.
c) Selection of the conformity assessment procedure
Fig. 1 shows which conformity assessment procedure manufacturers may choose depending on the class of the medical device.
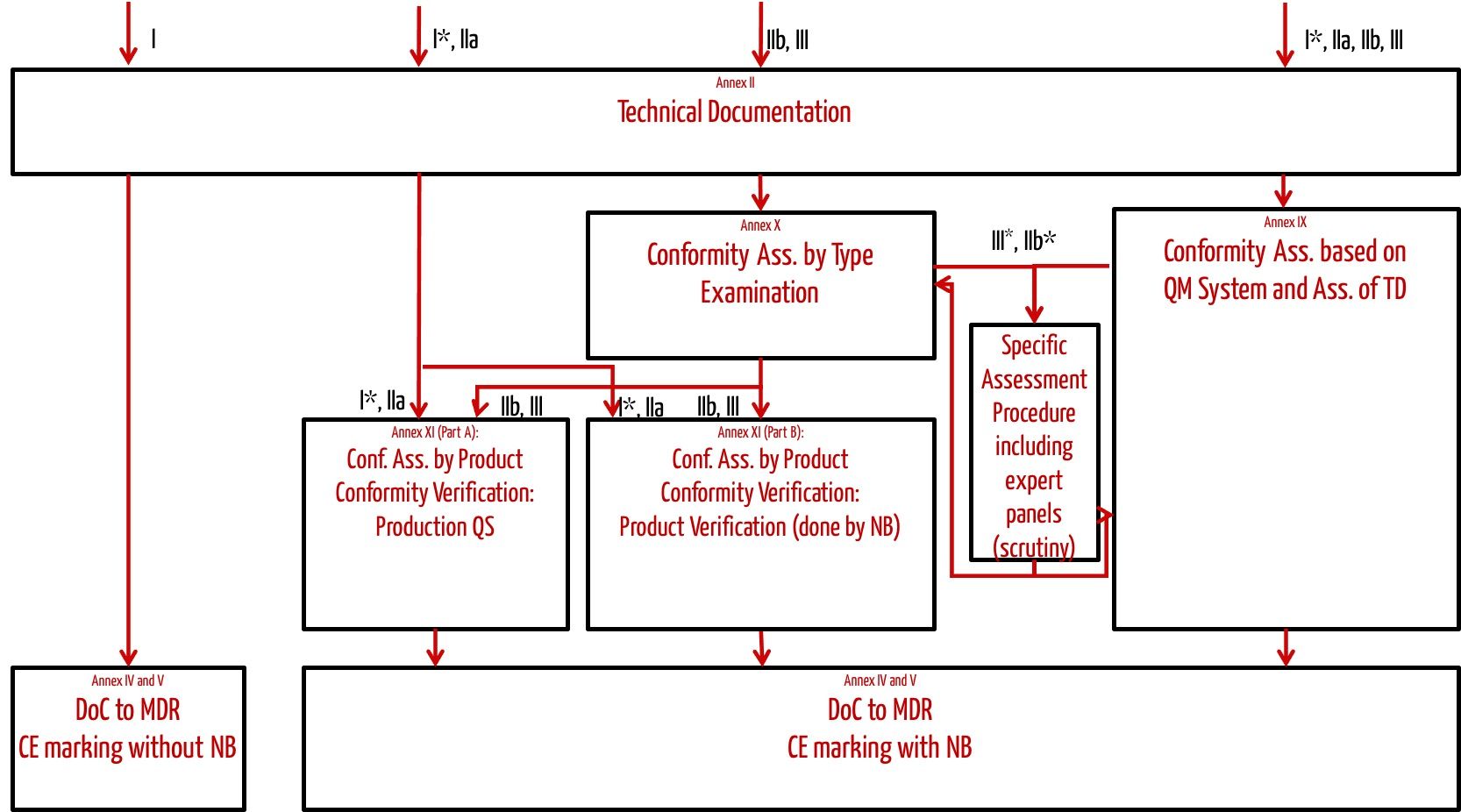
Fig. 1: Conformity assessment procedures specified by the MDR
The most frequently used conformity assessment procedure is the one according to Annex IX of the MDR, which requires the manufacturer to have a certified(!) quality management system.
2. Course of the conformity assessment procedures
The conformity assessment procedure leads to the CE mark. Manufacturers get there in seven steps:
- Determine the intended purpose of the device
- Determine applicable EU regulation (MDR, IVDR) and other regulatory requirements (including harmonized standards and MDCG guidelines)
- Determine the class of the device
- Select a suitable conformity assessment procedure
- Establish a QM system
- Develop the device according to QM system and regulatory requirements
- Declare conformity and affix CE mark
The following presentation shows these steps.

Fig. 2: Presentation “Conformity assessment procedure: 7 steps to the CE mark”. Click to open
Further information
Another article outlines the process from the initial idea to the device launched on the market.
3. Further articles on conformity assessment
a) Basics
The term conformity assessment is often equated with the terms approval and certification, but this is not correct:
- Approval of medical devices: Approval procedures in the EU and the USA
- MDR / IVDR certification: Why the terms are incorrect
b) Proof of conformity
To prove that a device meets the general safety and performance requirements and is therefore compliant, manufacturers should use:
Manufacturers need a certificate if a notified body is involved in the conformity assessment. The article “Request versus application for certification” is helpful.
c) Specific conformity assessment procedures
Further articles concern specific conformity assessment procedures:
- Scrutiny procedures: Consultation procedure
- Type examination for software – is it possible?
d) Particularities
Some articles are relevant for individual product classes:
- Accessories for a medical device: Definition and regulatory requirements
- Hospital information systems as systems and procedure packs
- IVD (in vitro diagnostics)
- In-house manufacture of medical devices
- Combination products: Applicable law and regulatory requirements
4. Support throughout the procedure
a) Free micro-consulting
Do you have questions about conformity assessment? Then, benefit from our free micro-consulting.
b) Consultation
The experts at the Johner Institute specialize in supporting manufacturers in quickly bringing their medical devices to market and in compliance with the law. From the initial idea to the QM audit to the CE mark:
c) Online self-study courses
The Medical Device University is recommended for those who prefer to take the path largely without outside help. On this e-learning platform, you will learn how to complete all the above tasks step by step using customized video training. You will also receive a complete set of templates and sample templates.
Contact us now to find the quickest way to obtain a CE mark for your medical device.
Contact us