Qualification and classification are mandatory tasks of manufacturers, notified bodies, and authorities to determine the regulatory requirements for medical devices.
Content
This page provides an overview and links to relevant articles:
- Articles on qualification and classification
- Further articles on this topic
- Support for qualification and classification
1. Articles on qualification and classification
a) Overview
Caution!
The term “classification” is used several times for medical devices. In addition, “classification” is often used when referring to a qualification.
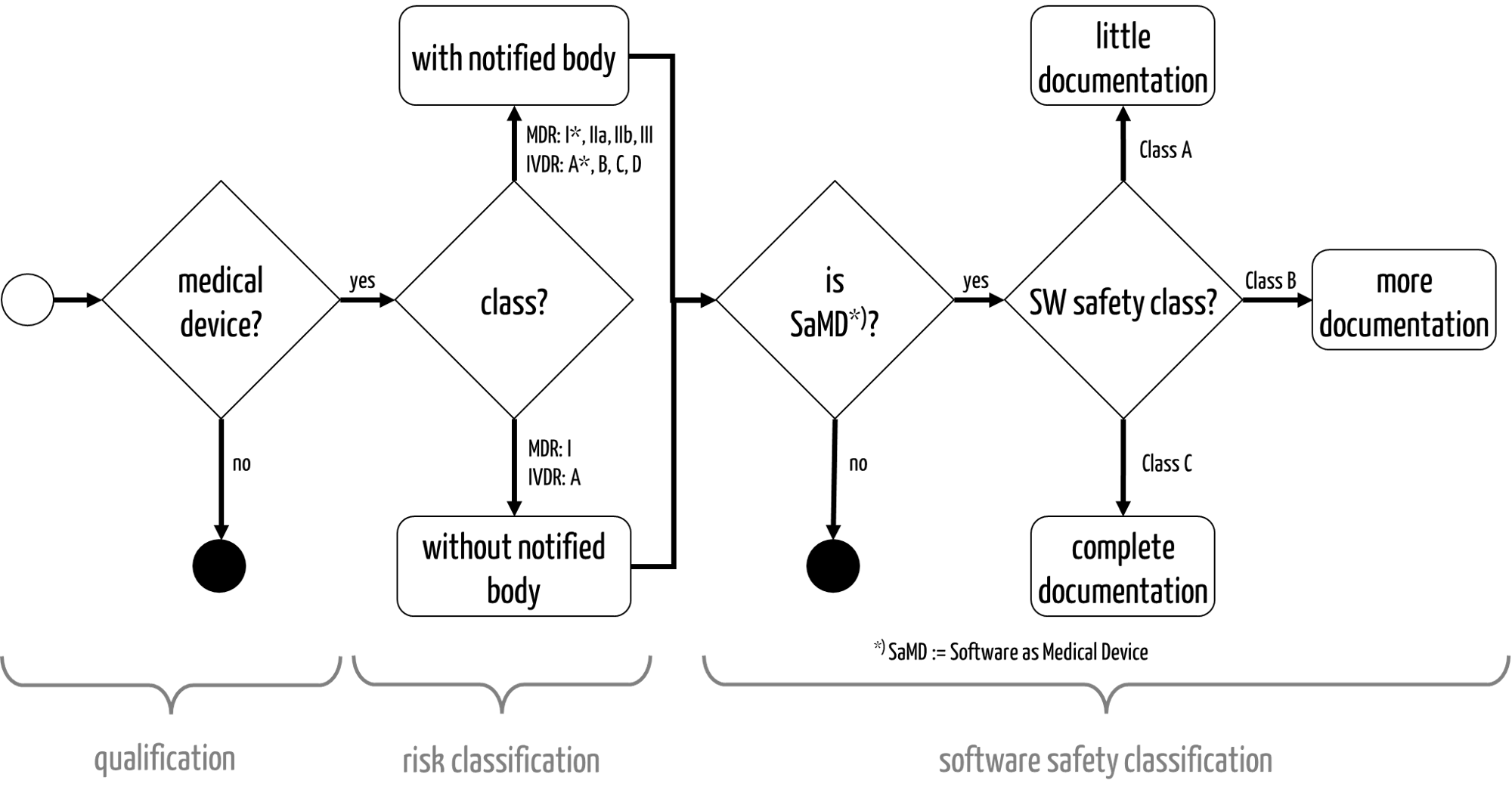
Fig. 1: The qualification and classification of medical devices
b) Qualification as a medical device
The first “classification” is qualification. This is the decision whether a device is a medical device (or accessory) or not.
The EU MDR and IVDR qualify all devices as medical devices (comparable to the FDA) – in simple terms – if the manufacturer intends them to diagnose, monitor, alleviate, predict, and treat illnesses and injuries in patients.
The exact definition can be found in the MDR in Article 2, paragraph 1.
This decision is almost always based on the intended (medical) purpose. According to MDCG 2019-11, functionality also plays a role in the case of software.
Examples of medical devices
Medical devices include, among others:
- Medical electrical equipment such as X-ray machines, CTs, MRI devices, ultrasound devices, electrical blood pressure monitors
- Surgical instruments, e.g. scalpels, forceps, tweezers
- Hip implants
- Many aids, e.g., wheelchairs and bandages
- Some software applications, such as DiGA, PDMS, and RIS
Examples of products that do not count as medical devices
The following are not considered medical devices
- Hospital information systems that are only used for documentation purposes
- Most wellness and fitness devices
- Medicinal products
- Devices that were not specifically marketed by the manufacturer for use on patients but can be used for this purpose
The “classifications” are also part of this qualification:
Caution!
For many devices, it is an important strategic decision as to which parts of a system the manufacturer places on the market as one or more medical devices or, as an accessory for a medical device or as a non-medical device. The Johner Institute can help with this (see below).
c) Classification of medical devices (“risk class”)
Overview: How is the risk class of a medical device determined?
How the risk class of a medical device is determined depends on the legal jurisdiction:
- In the EU and many other countries, there are explicit classification rules (e.g., in the MDR in Annex VIII).
- The FDA, on the other hand, explicitly defines the risk class for each product category. If a product cannot yet be assigned to a category, this must be applied for.
The risk classification determines the possible conformity assessment or authorization procedures (see Fig. 1).
- The MDR distinguishes between class I, I*, IIa, IIb, and III.
- The IVDR distinguishes between classes A, B, C, and D.
- The FDA divides devices into classes I, II, and III.
In Europe only devices in the lowest “risk class” do not require notified bodies to be involved in the conformity assessment procedure.
Further information
Rule 11 for medical devices that contain or are software leads to many discussions in the EU.
Part of this classification is the categorization:
- Medical device with or without a measuring function
- Medical device that counts as a reusable surgical instrument
- Sterile medical device
Devices that fall into one of these classes and would otherwise be assigned to class I are classified as class I*.
c) Software safety classification
Finally, there is a classification specifically for software, whether it is “software as a medical device” (standalone software) or software as part of a medical device:
- IEC 62304 distinguishes between software safety classes A, B, and C. These affect the scope of the software documentation to be created (see Fig. 1).
- On the other hand, the Levels of Concern (now “Documentation Levels”) determine the type of software documentation to be submitted.
2. Further articles
Many other articles are relevant for the qualification and classification of medical devices:
- For some devices, the decisive factor for the class is whether they diagnose or influence vital physiological processes.
- Please also refer to the articles on class I medical devices (in general) and class I software (in particular).
- When parameterizing software, operators should take care not to create or modify a medical device.
- In this context, the impact of MDR and IVDR on health institutions such as clinics and other operators is also relevant.
- Software as a medical device poses particular challenges.
- The IMDRF (International Medical Device Regulators Forum) guides qualification and classification.
The article “Classification: 8 Tips for Precise Pigeonholing” provides overarching considerations for classifying concepts.
3. Support with qualification and classification
Do you still have questions about the qualification or classification of medical devices? Then, please take advantage of our free micro-consulting service.
The experts at the Johner Institute specialize in developing a regulatory strategy that will enable you as a manufacturer to bring your devices to market worldwide as quickly and flexibly as possible. Contact us right here.