The European legislation defines systems and procedure packs and distinguishes between different configurations. The regulatory requirements placed on the manufacturer are heavily dependent on these configurations.
This article will explain to you what legislators understand by systems and procedure packs, what the most important legal requirements are for manufacturers, and what common mistakes you should avoid.
1. Systems and procedure packs: what are they?
a) Definitions
Both the German Medical Device Act (MPG) and the EU’s Medical Device Directive (MDD) set requirements for the marketing of systems and procedure packs. But neither defines the terms. The EU Medical Device Regulation (MDR) – not to be confused with the German Medical Device Regulation (MPV) – makes up for this omission:
“System” means a combination of products, either packaged together or not, which are intended to be inter-connected or combined to achieve a specific medical purpose;
Source: MDR
The MDR also defines the term “procedure pack”.
“Procedure pack” means a combination of devices packaged and marketed together and intended for use for a specific medical purpose;
Source: MDR
The definitions make it clear that there is an overlap between systems and procedure packs (see Fig. 1).
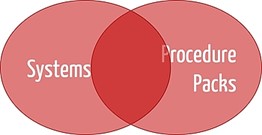
b) Examples of systems and procedure packs
Examples of combinations of products that are systems and procedure packs:
- Electrosurgery devices marketed in a pack together with high-frequency electrodes
- Implants marketed in a pack together with bone cement
- Syringe pumps marketed in a pack together with disposable syringes
Examples of combinations of products that are systems but not procedure packs:
- All of the above examples if the products are sold individually but should be/are combined (in line with the intended purpose)
Examples of combinations of products that are procedure packs but not systems:
- Blood pressure monitor marketed in a pack together with a suitable disinfectant
- Infusion pumps (medical device) and software for controlling infusion pump parameters (accessory) that are marketed together in one pack
c) Are hospital information systems systems or procedure packs?
A Court of Justice of the European Union (CJEU) decision ruled that Hospital Information Systems (HIS) can be non-medical devices but may contain modules that are classified as medical devices. In this context, we are then faced with the question of whether such an HIS is a system or procedure pack according to the legal definition.
A detailed expert opinion analyses whether hospital information systems should be considered as systems or procedure packs (German).
d) Difference to combination products
Combinations of medical devices and medicinal products or biological products are considered “combination products” and do not fall under the definition of “systems and procedure packs”.
The article on combination products describes the combination of medical devices and medicinal products and the regulatory requirements.
e) Difference to systems according to IEC 60601-1
IEC 60601-1 also talks about systems, or more precisely “medical electrical systems” (ME systems, for short). It defines these as follows.
“Combination, as specified by its manufacturer, of items of equipment, at least one of which is a medical electrical system to be inter-connected by functional connection or by use of a multiple socket-outlet”
Source: IEC 60601-1, Clause 3.64
In contrast to the definition of systems in the MDR, medical electrical systems are not necessarily supplied by the manufacturer as “systems”. Instead, it is often the operators who assemble the ME systems.
The manufacturer can deliver an ME system as a medical device comprising several “components” or as a system as defined by the Medical Device Regulation (MDR).
f) What are not systems and procedure packs
The MDCG has published the guideline MDCG 2018-03 (here is the link to MDCG 2018-03 rev. 1). In this guideline, the MDCG provides guidance on the assignment of UDIs for systems and procedure packs. However, it also determines what it does not consider to be a system and procedure pack:
Based on a request of a client or hospital, a natural or legal person in the supply chain may make available together different products, including CE marked devices, which are – in that entire combination – neither placed on the market by that natural or legal person, nor intended by that natural or legal person to be used together for a specific medical purpose. Devices made available in the described manner are not considered as systems or procedure packs.
MDCG 2018-03
This means that the decision as to whether the combined placing on the market of products results in them being understood as a system or procedure pack also depends on whether the ” system or procedure pack producer ” is also the marketer of one of the individual devices in this system or procedure pack.
Obviously, the EU wants to achieve simplification for companies that “only” place devices on the market, e.g., as “convenience kits.” As an example, the MDCG cites:
Example: a distributor supplies, upon request of a client, in one shipment, sterile tweezers, a sterile needle and surgery gloves.
MDCG 2018-03
2. Which regulatory requirements must be followed?
European jurisprudence consists of, firstly, the medical device directives that have to be transposed into national (medical device) law, and the EU medical device regulations (MDR, IVDR). This section describes the regulatory requirements that manufacturers who market systems or procedure packs must comply with.
- Medical Device Directive (MDD)
- German Medical Device Act (MPG)
- German Medical Device Regulation (MPV)
- EK-Med
- EU Medical Device Regulation (MDR)
a) Medical Device Directive
In Article 12, the Medical Device Directive (MDD) requires manufacturers of systems and procedure packs to draw up a declaration by which it states that:
he has verified the mutual compatibility of the devices in accordance with the manufacturers’ instructions and has carried out his operations in accordance with these instructions; and he has packaged the system or procedure pack and supplied relevant information to users incorporating relevant instructions from the manufacturers; and the whole activity is subjected to appropriate methods of internal control and inspection.
The MDD did not talk about manufacturers. The fact that the requirements apply indirectly to manufacturers is a result of the German transposition of these requirements into MPG §10(1) and MPV §7(6). Only the latter talks about the manufacturer who has to issue the declaration.
Manufacturers do not have to carry out (another) conformity assessment for these systems and procedure packs. However, the legislation presupposes that the products put together will bear CE marking and will be put together and used “within their intended purpose”.
The MDD defined “devices” as follows:
“Both medical devices and accessories shall hereinafter be termed devices.”
Source: MDD, Article 1
Therefore, the MDD only referred to medical devices and accessories, not IVDs.
As soon as the combination contains a device that is not used according to the intended purpose or one that has not been marketed in accordance with the MDD, this combination (without a conformity assessment procedure) may not be marketed as a system or procedure pack.
Manufacturers had to ensure that the declarations are “kept at the disposal of competent authorities for a period of five years”. The German Medical Device Act with its national regulations regulates this in more detail.
It seems that the authorities themselves were not sure when such a declaration had to be made. The head of a regional council wrote to one of the Johner Institute’s clients to say:
We have re-examined the process and, in consultation with the other regional councils, we have decided not to accept an additional notification as a system assembler for the manufacturer declaration.
The reason for this is that the compatibility of the accessory software with the main software must already have been proven as part of the conformity assessment procedure. We therefore consider a declaration as the party responsible for assembling systems or procedure packs necessary only if you are not the manufacturer as defined by the MPG of at least one medical device in the system.
The legal basis for this assessment is not clear from the text. However, this loose interpretation seems to be conclusive and should be in the minds of a lot of manufacturers.
b) German Medical Device Act
The German Medical Device Act transposed Article 12 of the MDD into German law through § 10. The relevant part of the law states:
(1) Medizinprodukte, die eine CE-Kennzeichnung tragen und die entsprechend ihrer Zweckbestimmung innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen zusammengesetzt werden, um in Form eines Systems oder einer Behandlungseinheit erstmalig in den Verkehr gebracht zu werden, müssen keinem Konformitätsbewertungsverfahren unterzogen werden. Wer für die Zusammensetzung des Systems oder der Behandlungseinheit verantwortlich ist, muss in diesem Fall eine Erklärung nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1 abgeben.
In Paragraph 4, the MPG prohibited additional CE markings being affixed to systems and procedure packs, and required that necessary information be provided by the marketer of the system or procedure pack.
The MPG also stressed that a conformity assessment procedure for a system or procedure pack could only be omitted if the system or procedure pack only includes medical devices used in a way that is compatible with the original intended purpose.
The MPG firmed up this obligation to notify the authorities in § 25 “General obligation to notify”.
(2) Wer Systeme oder Behandlungseinheiten nach § 10 Abs. 1 zusammensetzt oder diese sowie Medizinprodukte nach § 10 Abs. 3 sterilisiert und seinen Sitz in Deutschland hat, hat der zuständigen Behörde unter Angabe seiner Anschrift vor Aufnahme der Tätigkeit die Bezeichnung sowie bei Systemen oder Behandlungseinheiten die Beschreibung der betreffenden Medizinprodukte anzuzeigen.
Accordingly, those responsible for putting together systems or procedure packs, i.e., assemblers, had to notify the competent authority.
In other words, manufacturers had to notify the German Institute of Medical Documentation and Information (DIMDI), which then forwarded the notification to the state authority responsible for registration.
We have had it confirmed to us by the Trade Supervisory Board of the Government of the Upper Palatinate that manufacturers have to report systems and procedure packs to the DIMDI via its website.
c) German Medical Device Ordinance (MPV)
The aforementioned § 10 MPG referred to the corresponding legal regulation according to § 37 of the law. This legal regulation was the Medical Device Ordinance (MPV) in the version dated 9/27/2016, including § 7 “Conformity assessment procedure for other Medical Devices”.
(6) Für Systeme und Behandlungseinheiten nach § 10 Abs. 1 des Medizinproduktegesetzes hat der Hersteller die Erklärung nach Artikel 12 Abs. 2 Satz 1 der Richtlinie 93/42/EWG auszustellen. Die Erklärung ist mindestens fünf Jahre und im Falle von implantierbaren Produkten mindestens 15 Jahre aufzubewahren. Für Systeme und Behandlungseinheiten nach § 10 Abs. 2 des Medizinproduktegesetzes gelten die Vorschriften der Absätze 1 bis 4 entsprechend.
d) EK-Med
EK-Med also discussed the topic of systems and procedure packs, for example in Document 3.10 A 15 (German). It discussed what happens if the devices all have CE marking and are used for their intended purpose, but procedures are used (e.g., during packaging) that influence the product quality.
e) EU Medical Device Regulation (MDR)
The European Medical Device Regulation (MDR) took the concepts in the MDD further. It contains Article 22 “Systems and procedure packs” and Article 29 “Registration of products”. The MDR also regulates how to process the UDI.
Article 22 “Systems and procedure packs” paragraph (1)
Article 22 of the MDR corresponds to Article 12 of the MDD. However, in Paragraph (1) c) it opens the door to “other devices” and for in vitro diagnostics.
Natural or legal persons shall draw up a statement if they combine devices bearing a CE marking with the following other devices or products, in a manner that is compatible with the intended purpose of the devices or other products and within the limits of use specified by their manufacturers, in order to place them on the market as a system or procedure pack:
1. Other devices bearing the CE marking;
2. In vitro diagnostic medical devices bearing the CE marking in conformity with Regulation (EU) 2017/746;
3. Other products which are in conformity with legislation that applies to those products only where they are used within a medical procedure or their presence in the system or procedure pack is otherwise justified.
MDR Article 22, Paragraph 1c
The term “other devices or other products” is to be understood differently in 1. and 3. In 1. a device, according to Article 1, Paragraph 4, is a medical device, an accessory, or a device, according to Annex XVI, that must comply with the requirements of the MDR.
In 3. the term “other products” “device” refers to a product that is not subject to the requirements of the MDR, but must comply with other regulatory requirements and therefore may also bear a CE marking.
So it makes clear that non-medical devices as well as medical devices can be placed on the market as a system or procedure pack.
Non-medical devices must comply with applicable (European) legislation. Examples of such legislation include EU regulations and EU directives, such as REACH (1907/2006/EC) and the Radio Equipment Directive (2014/53/EU).
Article 22 “Systems and procedure packs” paragraph (2)
Paragraph (2) also requires the declaration, which must now be somewhat more precise and with which the manufacturer must confirm that it has provided the necessary information and has carried out appropriate monitoring, verification, and validation.
In the statement made pursuant to paragraph 1 the natural or legal person concerned shall declare that:
a) They verified the mutual compatibility of the devices and, if applicable other products, in accordance with the manufacturers’ instructions and have carried out their activities in accordance with those instructions;
b) They packaged the system or procedure pack and supplied relevant information to users incorporating the information to be supplied by the manufacturers of the devices or other products which have been put together;
c) The activity of combining devices and, if applicable, other products as a system or procedure pack was subject to appropriate methods of internal monitoring, verification and validation.
The MDR requirement in Article 22(2)(3) on “Product verification and validation” goes further than the MDD requirements in Article 12(2)(c). According to the MDD, the manufacturer “only” has to confirm that “the whole activity is subjected to appropriate methods of internal control and inspection”.
Verification and validation implicitly requires specifications that can be used by the manufacturer for verification and validation. This in turn requires a specification for the “combined” system. The MDD does not contain such (indirect) requirement.
Article 22 “Systems and procedure packs” paragraph (4)
Paragraph (4) mentions the term “devices” again:
Where the system or procedure pack incorporates devices which do not bear the CE marking or where the chosen combination of devices is not compatible in view of their original intended purpose, or where the sterilisation has not been carried out in accordance with the manufacturer’s instructions, the system or procedure pack shall be treated as a device in its own right and shall be subject to the relevant conformity assessment procedure pursuant to Article 52. The natural or legal person shall assume the obligations incumbent on manufacturers.
The MDD required a new conformity assessment for systems or procedure packs that included:
- Medical devices bearing a CE marking, and
- Non-medical devices or/and
- Medical devices that are not used for their original intended purpose
On the other hand, the MDR requires a new conformity assessment for systems or procedure packs that include:
- Medical devices bearing a CE marking, and
- Medical devices that do not bear a CE marking (this could be, e.g., an FDA-approved medical device) or/and
- Medical devices that are not used for their original intended purpose
In contrast to the MDD (as shown above), the MDR no longer directly rules out the combination of a medical device with a non-medical device without a new conformity assessment.
Article 29 “Registration of devices”
An additional requirement contained in the MDR compared to the MDD is that manufacturers must assign a UDI to the system or procedure pack and enter it in the EUDAMED:
2. Before placing on the market a system or procedure pack pursuant to Article 22(1) and (3), that is not a custom-made device, the natural or legal person responsible shall assign to the system or procedure pack, in compliance with the rules of the issuing entity, a Basic UDI-DI and shall provide it to the UDI database together with the other core data elements referred to in Part B of Annex VI related to that system or procedure pack.
Annex VI describes these requirements in more detail.
UDI: Annex VI Part C
The MDR defines the Unique Device Identification (UDI) in Paragraph 3.7 of Annex VI:
“Systems and procedure packs as referred to in Article 22 shall be assigned and bear their own UDI”.
In 6.3.1, it specifies:
“The natural or legal person referred to in Article 22 shall be responsible for identifying the system or procedure pack with a UDI including both UDI-DI and UDI-PI.”
Finally, the MDR also regulates where the UDI has to be applied:
6.3.2. Device contents of system or procedure packs shall bear a UDI carrier on their packaging or on the device itself.
Exeptions:
1. Individual single-use disposable devices, the uses of which are generally known to the persons by whom they are intended to be used, which are contained within a system or procedure pack, and which are not intended for individual use outside the context of the system or procedure pack, shall not be required to bear their own UDI carrier;
2. devices that are exempted from bearing a UDI carrier on the relevant level of packaging shall not be required to bear a UDI carrier when included within a system or procedure pack.
6.3.3. Placement of the UDI carrier on systems or procedure packs
1. The system or procedure pack UDI carrier shall as a general rule be affixed to the outside of the packaging.
2. The UDI carrier shall be readable, or, in the case of AIDC, scannable, whether placed on the outside of the packaging of the system or procedure pack or inside transparent packaging.
MDR, Annex VI, Paragraph 6.3.2
Summary of changes made by the MDR
The main changes introduced by the MDR compared to the MDD are:
- Systems and procedure packs can include non-medical devices.
- Manufacturers must confirm that they have verified and validated such “combinations”, which in turn requires, among other things, specifications that can be used for the verification and validation.
- Systems and procedure packs require their own UDI.
f) FDA requirements for systems and procedure packs
The FDA still uses the term “convenience kits”:
“Convenience kit means two or more different medical devices packaged together for the convenience of the user.”
Source: FDA 21 CFR 801.3
This is a bit like a procedure pack, although the definition of a procedure pack does not specify whether the packaging together of the medical devices is for convenience or not. The devices packaged together in a convenience kit are not necessarily (all) used for a single medical purpose.
Examples of convenience kits are first aid kits or sets of devices that have been packaged together for a particular operation. The FDA published the requirements for the assignment of a UDI to these kits in its “UDI Convenience Kits” guidance document.
The FDA still also uses the term “device packages”:
„Device package means a package that contains a fixed quantity of a particular version or model of a device.“
Source: FDA 21 CFR 801.3
However, this does not correspond to the systems and procedure packs as defined in the European legislation.
3. Summary and conclusion
a) Regulatory requirements
Compared to the MDD, the MDR has simplified the requirements in the sense that systems and procedure packs may contain non-medical devices. These must be verified and validated by the manufacturers in accordance with the MDR; this was not required so explicitly by the MDD.
Unlike the MDD, the MDR does not need to be transposed into national law. When asked how well the MDD succeeded in doing this, Dr. Banz came to the conclusion that:
“[…] it is almost impossible to fulfill the requirements and overcome the hurdles created by the literal application of §10(2). These difficulties, together with the absence of any justification by the legislative authorities for going beyond the framework established in Article 12(2), sentence 2 of the MDD, lead to the conclusion that the directive may after all be imprecisely implemented in national law.”
You can read the full article by Banz/Eckle/Kremser on “Systemen und Behandlungseinheiten und Ihre Behandlung nach dem Medizinprodukterecht” [Systems and procedure packs and how to handle them in accordance with medical device law] online and in MPR2/2009 p. 50ff.
The fact that the authorities themselves disagree on when a declaration has to be made (see above) regarding a system or procedure pack creates further legal uncertainty.
b) Unawereness of regulatory requirements
A lot of manufacturers are unaware of the regulatory requirements they must meet if they want to sell systems and procedure packs. Armin Gärtner writes to us:
“Regulatory authorities are increasingly demanding Article 12 declarations in hospitals, e.g. for endoscopy equipment trolleys and for other systems supplied by a manufacturer. Unfortunately, a lot of manufacturers are simply unaware of this. Others shift the responsibility for putting together products sold individually onto hospitals, which usually do not know or understand this.”
c) Limitations
This article has not discussed combination products (medical devices and pharmaceutical products). Nor has it looked at when it makes more sense to market devices individually, as systems or procedure packs or as one device.
Read a separate article for a discussion of the pros and cons of combining medical devices.
Change history
- 2024-08-06: Reference in 1.d) added. Chapter 2.a) to 2.d) reworded in past tense
- 2020-03-18: First version of the article



Thank you for this article, very interesting to read.
You state:
“In contrast to the MDD […], the MDR no longer directly rules out the combination of a medical device with a non-medical device without a new conformity assessment.”
For me it is hard to follow this.
Paragraph (4) states “Where the system or procedure pack incorporates devices which do not bear the CE marking […] the system or procedure pack shall be treated as a device in its own right and shall be subject to the relevant conformity assessment procedure.” – typically, non-medical devices do not bear the CE marking, thus I would expect that the combination of a medical device with a non-medical device requires a new conformity assessment. Exception would be that the incorporated device would be a medical device, assessed e.g. by FDA or covered by other regulations.
Can you more explicitly describe how you get to your conclusion?
Thank you!
Dear Mr. Saborowski,
Thank you for your question! I will pass on my colleague’s reply below:
With CE marking, a distinction must be made between products that have a CE but are not for medical devices, such as a PC, and products that do not have an EU directive CE, such as FDA products. For the combination between a medical device with CE and a non-medical device with a CE, only the combination must be assessed. For the combination between a medical device with CE and a FDA product, the conformity assessment must be carried out in accordance with the MDR. This means that products from third countries do not have access to the EU market per se.
Best regards,
Mario Klessascheck
I hope this answer helps you.
Best regards,
Tea Bodrusic
Thank you for the excellent article.
If a procedure pack follows all the rules required to adhere to Article 12 (i.e., all products that require CE Mark conform; all products are used as per intended use; all products are compatible), what are the administrative requirements for such a PP. As a 3rd country, we have registered as a PP producer.
1. Do we require a Medical Device Importer to import PP into EU?
2. Do we require an Authorized Representative to import PP into EU?
3. Are all member states in EU aligned with the MDR/Article 22 on PP?
Dear Mrs Whalen,
If parts of the PP have not yet been placed on the EU market, you need an importer and an authorized representative. All member states are aligned with Article 22.
Best regards, Mario
Dear Mario,
I would also intuitively say that a producer located outside the Union would need an authorised representative. However, the truth is that I cannot find any mention of an authorised representative in Article 22, so I question what is the legal basis in the MDR for a producer (as opposed to a manufacturer) having to appoint an authorised representative?
Kind regards
Guten Tag, ich habe eine Frage, wenn ich ein Bett als MD verkaufe und die Seitenteile dazu, entweder zusammen mit dem Bett oder auch einzeln verkauft werden können, sind diese Seitenteile dann MDs oder Zubehöre? Als MD mit Bett ist es in dem Fall ja ein System, richtig? Bekommen diese Seitenteile dann auch eine extra UDI-DI?
Danke
MFG
Liebe Frau Frank,
ich darf Ihnen folgende Antwort von unserem Experten weiterleiten:
Liebe Frau Frank,
Ausschlaggebend ist die Zweckbestimmung. Für Zubehör gilt, dass es die klinische Funktion des Medizinprodukts gezielt und unmittelbar unterstützt oder überhaupt erst ermöglicht. Die Seitenteile können somit als Zubehör eingestuft werden und erhalten eine eigene UDI. In der MDR wird Zubehör wie ein eigenständiges Medizinprodukt behandelt, was bedeutet, dass für es alle erforderlichen Aktivitäten, einschließlich einer klinischen Bewertung, durchgeführt werden müssen. Dabei beziehen sich die Endpunkte vor allem auf die Sicherheit, weniger auf die klinische Leistung. Außerdem kann die technische Dokumentation für das Zubehör zusammen mit der des Bettes in einer Akte geführt werden.
Liebe Grüsse, Mario Klessascheck
——
Herzliche Grüße
Tea Bodrusic
Thank you for a very informative article!
I’m a bit confused about how this works in context of IEC 60601-1 though.
If I combine a medical electrical device (ME Equipment) with e.g. a standard PC (CE marked) through some electrical connection, then I have a ME System according to 60601-1 and I would expect that clause 16 of the standard comes into play.
This, I believe, makes sense since combining two pieces of electrical equipment could e.g. cause altered leakage currents. Yet it seems like 60601-1 can be left completely out of the equation. Is this true or am I missing something?
Best regards
Jesper
Hello, Jesper,
I think that this would not constitute a “Procedure Pack or System” necessarily. The computer hook-up presumably provides software element that is part of the medical device, and not an independent component that can be bundled and is compatible with the medical device. The question is whether the software is a medical device in its own right and independent of the medical device? Or is the function of the medical device dependent on the software?
I would be interested to understand how Article 11 distinguishes these types of bundled products.
Thanks for your reply Anne Marie.
So if we say that the external PC, with some software on it, is an extension of the medical electrical device and provides extra features for that specific device, then this combination should be viewed as a medical device on its own and is therefore not a “system” in the eyes of MDR article 22?
Best regards
Jesper
Hi Jesper,
MDCG 2023-4 “Medical Device Software (MDSW) –Hardware combinations: Guidance on MDSW intended to work in combination with hardware or hardware components” (October 2023) addresses the multiple possibilities for SW-HW combinations under EU-MDR. Compliance to Article 22 is one of at least 3 options that need to be considered.
Dear Jasper,
This assumption is correct. Since your technical file covers the entire ME system as well as EMC and safety tests, a declaration under Article 22 would not be required.
best regards, Mario
Dear Jasper,
You are right, this would be an ME system, which should not be confused with the term system in Article 22 MDR. The requirements are different. If you place the ME system on the market, you do not need an Article 22 declaration for this type of system. Article 22 refers more to combinations where you combine certified devices where you are not the manufacturer.
Thanks for your reply Mario!
Does this mean that if I place a system consisting of e.g. three different certified medical electrical devices – all electrically interconnected and each with its own supply mains connection – on the marked, then I can do so based on an Article 22 declaration without ever employing 60601-1?
Best regards
Jesper
Dear Jasper,
The concept of a “system” differs depending on the context:
– According to IEC 60601-1, the system definition is based on technical risk considerations. You must assess the impact on basic safety and essential performance, particularly in accordance with Clause 16 and other relevant clauses. Additionally, complete technical documentation for the medical electrical (ME) system must be prepared.
– According to Article 22 of the MDR, the system concept has a regulatory background. A declaration under Article 22 is required only when a manufacturer assembles different CE-marked devices or components into a new combination with a specific medical purpose, and places this combination on the market.
If you are the original manufacturer of the entire ME system, and the system remains under your control (i.e., you define and market it as a single product), then no declaration under Article 22 is needed.
An example of when Article 22 applies:
A manufacturer—or even a distributor—combines separate CE-marked components from other manufacturers (e.g., a trolley, an endoscopy device, a monitor, and a printer) to build a mobile endoscopy station. If this combination is then placed on the market as a new system for a specific medical purpose, it falls under Article 22 and requires a formal declaration.
Best regards, Mario
Apologies…Article 22.
Guten Tag,
ist es möglich ein MDR procedure pack (label des PP entspricht den MDR Anforderungen, PP selbst besitzt keine eigene IFU, da alle IFUs der einzelnen Komponenten enthalten sind) auf den Markt zu bringen, auch wenn alle enthaltenen Medical Device Komponenten noch unter der MDD (labels, IFUs etc. nach MDD) laufen?
Herzlichen Dank und beste Grüße!
Liebe Frau Jung,
Ich antworte auf Deutsch, da Sie Ihre Frage ebenfalls auf Deutsch gestellt haben.
Zu Ihrer Frage: Die MDCG hat dies in einer entsprechenden Guidance MDCG 2021-25 Rev. 1 bereits geklärt.
Wenn Sie vor dem 26. Mai 2021 unter der MDD eine Artikel-12-Erklärung erstellt haben und die betroffenen Produkte unter die Übergangsregelung gemäß Artikel 120(3) der MDR fallen, müssen Sie keine neue Artikel-22-Erklärung nach MDR ausstellen. Die Artikel-12-Erklärung bleibt gültig, bis die Übergangsfrist für eines der enthaltenen Produkte abläuft.
Hilft Ihnen das weiter?
Herzliche Grüsse, Mario Klessascheck
If I want to place a medical device combined with a non medical CE marked product on the market in a System under the MDR what is than this justification which they are looking for (MDR Article 22, Paragraph 1c)?
Dear JO,
The “appropriate justification” under MDR Article 22(1)(c) is a documented rationale proving that the combination of a medical device with a non-medical CE-marked product is safe, functional, and suitable for its intended use, without altering the intended purpose or creating new regulatory obligations.
The justification is a technical and regulatory rationale showing that:
– A clear description of the combination (e.g., medical device + CE-marked accessory)
– Declaration of conformity of the non-medical CE product to its applicable legislation
– A risk analysis of the combined use
– Evidence of compatibility (mechanical, electrical, functional)
– Confirmation that the intended use of the system remains within that of the medical device
– If needed: Instructions for safe combination and use
The justification should include a description of the combination, the non-medical product’s declaration of conformity, evidence of compatibility, confirmation that the intended use remains within that of the medical device, and, if necessary, instructions for safe use.
Best regards, Mario
Hi , what I find a bit confusing is your reference to manufacturers while this should be system pack producer, in my opinion. The role is important as this has consequences for the product label. In my view a system pack or procedure pack should not have a manufacturer symbol on the final label, but just clarify the adress of the system/procedure packer. Do you agree and adapt the term in you text?
Dear Erwin,
yes you are correct. The system pack producer is not necessarily the manufacturer. Sorry for the inaccurate use of terms.
best regards, Mario
Hi, If a system packer (SPPP) includes multiple CE devices in a system pack, of which are some from outside the union (making the SPPP also an importer), should it be sufficient to have identification of individual CE manufacturer’s name and adresses, as wel as the importer names and adressen be part of the accompanying documentation with the system pack and have this not all included on the product label. Or is it mandatory on the label?
Dear Erwin,
according to the MDCG Guidance (MDCG 2021-27 Rev.1) the importer’s details must be included on the device (or on its packaging, or in a document accompanying the device).
Best regards, Mario
In the text you refer to manufacturer, but this should be System pack or procedure pack producer (SPPP) under EU MDR. Am I correct? i ask as this has consequences. A manfacturer of system pack assumes responsibility as a manufacter, a system packer does not. Also a formal SPPP does not carry a manufacturer symbol on the label. Do you agree?
Dear Erwin,
Yes, I agree. See my comment on your first post.
Best regards, Mario