Manufacturers of (most) class III medical devices have to undergo the Premarket Approval (PMA) procedure. That is one of the FDA’s most complex submission pathways, with a typical processing time of more than one year. The FDA usually approves only a few dozen PMA applications per year.
This article presents the FDA’s requirements and tips on how manufacturers can overcome this high hurdle.
1. Premarket Approval (PMA): The basics
1.1 Application of the PMA procedure
The FDA requires a PMA, especially for class III medical devices. These are devices that are life-saving, prevent serious injury, or pose a significant health risk. Examples include heart valves, complex diagnostic tests, or life-supporting devices.
Check the FDA’s Product Classification Database to see if your device is already listed.
1.2 Regulatory framework
Title 21 Part 814 of the Code of Federal Regulations (CFR) describes the PMA procedure and specifies the requirements that manufacturers must fulfill in this procedure.
Additional regulatory requirements must be met, such as those for
- labeling (21 CFR Part 801),
- the quality management system (21 CFR Part 820),
- clinical Investigations (21 CFR Part 812).
1.3 Types of PMA procedures
The FDA distinguishes between several types of PMA procedures.
| Name of the PMA procedure | Description | Use, suitability |
| Original PMA | Procedure in which the manufacturer submits all the required documents to the FDA in one step | This type is generally used when the device has already been thoroughly tested and approved in other countries with existing requirements. |
| PDP (Product Development Protocol) | Procedure in which the development and testing of a device are combined to achieve market approval. The PDP is like a contract between a manufacturer and the FDA. It defines the necessary design and development activities, including specific milestones and acceptance criteria for the respective device. When the FDA recognizes a PDP as complete, it is considered an approved PMA, which means formal approval of the device for the market. | Well suited for devices with established and well-known technologies. |
| Modular PMA | This approach divides the approval procedure into individual, precisely defined sub-areas (modules). Each module is submitted to the FDA as soon as it is completed. This means that the procedure can be processed step by step over time. The advantage of this is that the FDA can provide feedback promptly. | The approach is particularly suitable for devices still in the early stages of clinical investigation. |
If you are unsure which type is right for you, discuss any open points with the FDA before submission, e.g., as part of a pre-submission procedure. The Johner Institute can support and guide you through the pre-submission process.
1.4 Duration and costs
For FY 2025, the costs for a PMA application are:
Standard fee: USD 540,783.00
Small business fee: USD 135,196.00
There are also fee waivers for pediatric devices or on first submissions for specific small businesses.
The FDA’s objective is to review PMA procedures within 180 days (decision with no committee input) or 320 days (decision with committee input).
2. Contents of a PMA application
A complete PMA application contains both administrative and technical documents.
2.1 Administrative documents
The administrative part of a PMA application includes:
- Signed cover letter with company and product details
- For international manufacturers: countersigned by the authorized US representative
- 10- to 15-page application summary with indications, device description, alternative treatment methods, marketing history, and an overview of the studies conducted
The following is also required:
- A detailed table of contents
- The financial certification or disclosures of the clinical investigators (if applicable)
Ensure your submission is clearly structured and includes a detailed table of contents. Use the FDA’s checklist to review the completeness of your submission or use the eSTAR template for applicable PMA types.
2.2 Technical documents
The technical documentation is the centerpiece of the PMA. The required contents can be found in 21 CFR Part 814.20. They provide complete scientific proof of safety and effectiveness and cover several areas.
2.2.1 Technical information
The technical information includes:
- Precise intended purpose: This must include a description of the disease or condition that the device is intended to diagnose, treat, prevent, cure, or alleviate, as well as a description of the patient group for which the device is intended.
- Complete device description: This includes a complete description of the device and its functional components or parts, references to applied standards, and an environmental impact assessment in accordance with 21 CFR 25.20(n).
- Complete design history file (DHF)
- Risk management documentation in accordance with ISO 14971
- Copies of labeling, e.g., accompanying materials
- QM information on processes and methods for manufacturing, processing, packaging, storage, and, if applicable, installation of the device, as well as the facilities and controls used for this
In most cases, the FDA also requires the submission of a representative sample of the device.
2.2.2 Scientific evidence
The scientific evidence includes:
- Preclinical study data
- Clinical study results
- Safety and effectiveness evidence
- Pediatric use data (if relevant)
The preclinical and clinical study data are at the heart of the PMA. They must provide scientifically sound evidence of the safety and effectiveness of the device in all relevant aspects. This includes:
- Preclinical tests (e.g., biocompatibility, mechanical & physical tests, V&V, or durability studies)
- Clinical experience
- Results of clinical investigations, including detailed study protocol, statistical analyses, patient data, safety and efficacy evidence, and adverse event documentation
- Benefit-risk assessment
3. Requirements for the QM system
3.1 Regulatory framework
A requirement for PMA submission is a quality management system (QMS) compliant with 21 CFR Part 820. In addition, it must meet the requirements of , for example, Part 803 (Medical Device Reporting) and Part 806 (Reports and Corrections).
Reason: The QM system must ensure that the medical device is manufactured in compliance with the PMA-specified requirements.
3.2 Inspection of the QM system
3.2.1 Submission of documents
The FDA requests selected documents from the QM system for the manufacture, processing, packaging, and storage of the devices with the PMA application. In addition, the FDA conducts a thorough on-site inspection.
3.2.2 Preapproval Inspection
The FDA may order a Preapproval Inspection during the review procedure. This inspection comprehensively reviews compliance with regulatory requirements, process quality, and documentation standards.
Particular attention is paid to process validation during the inspection, as it ensures the consistent safety and quality of the devices. At the time of the PMA application, the manufacturer may not yet have completed all the necessary process validations, so CDRH cannot review these. During the inspection, the manufacturer is expected to have all processes requiring validation fully validated. If it is determined during the inspection that this is not the case, the inspector may terminate the inspection.
3.2.3 Postapproval Inspection
The Postapproval Inspection takes place approximately 8 to 12 months after market approval and checks for continuous compliance with quality standards and change processes and the correct implementation of originally approved manufacturing and development specifications.
4. PMA procedure
4.1 Submission to the FDA
The PMA submission to the FDA is submitted electronically via the CDRH portal. It is sent either in eSTAR format (electronic structured application format with predefined fields) or as an eCopy (electronic copy of your application).
eSTAR format creation is voluntary for PMA applications and only possible for original PMAs. PDPs and modular PMAs must be submitted using the eCopy procedure.
4.2 The review procedure
A PMA is reviewed by an interdisciplinary team of 10 to 15 members. The review is a multi-step process involving three levels (regulatory, scientific, and quality management).

4.2.1 Acceptance Review
The Acceptance Review is an administrative process. It checks whether all the necessary documents are available. The applicant receives an e-mail within 15 days confirming whether the application has been accepted or not. In the latter case, the applicant receives a deviation list (not applicable to the eSTAR procedure).
4.2.2 Filing Review
The Filing Review reviews the contents to determine whether the application is sufficiently complete to be processed further. Within 45 days, the applicant will receive an e-mail notification stating whether the documents have been accepted for a detailed technical review or indicating missing information.
4.2.3 Substantive Review
During the Substantive Review, the FDA conducts an in-depth review of the application’s scientific, regulatory, and quality system aspects. During the review process, the FDA works closely with the applicant to resolve any identified deficiencies in a targeted and timely manner.
Make sure you have sufficient resources to answer any queries from the FDA promptly and in detail.
4.2.4 Substantive Interaction
During the Substantive Interaction phase, the FDA provides feedback to the applicant and decides on the further processing procedure:
a. Further processing of the PMA in an interactive review with the applicant OR
b. Issuance of a Major Deficiency Letter and postponement of the PMA until the deviations have been addressed
At this point, you have the option of holding a Day-100 Meeting. Furthermore, it is possible to submit a Q-Submission Request to discuss the submission’s progress or any deviations that have occurred.
You can request a Day 100 meeting with the FDA in your PMA application. Always request this meeting in your initial application! If you do not need it, you can cancel. If it should become necessary, you will save valuable time!
4.2.5 Approval decision
The FDA generally decides for or against approval after 180 days or 320 days. The following options are available:
a. Approval Order: The product can be sold.
b. Approval Pending Deficiencies Letter: The product cannot be sold. The decision addresses deviations that must be remedied before the PMA can be approved.
c. Approval Pending GMP Letter: The product cannot be sold. The QM system is not yet in compliance with FDA requirements.
d. Not-approvable Letter: The product cannot be sold. There are major deficiencies that may require new clinical or preclinical data.
The review period for the PMA submission depends on whether or not an advisory committee needs to be included. Without the involvement of a committee, the FDA aims to review PMAs within 180 days. With the involvement of the advisory committee, the planned time for the decision is extended to 320 days.
5. Post-approval requirements
After approval, the FDA may impose further requirements on the manufacturer, the fulfillment of which is crucial for market access.
5.1 Restrictions
These requirements may include restrictions on distribution and use, mandatory safety reports, and specific labeling and documentation requirements.
5.2 Studies
For certain product types, e.g., pediatric products, the FDA may order additional post-market surveillance studies (PMS studies).
5.3 Reports
Annual reports are a central element of post-approval monitoring and must document changes, scientific publications, and new findings regarding the safety and effectiveness of the medical device. The report enables the FDA to conduct a comprehensive risk assessment.
5.4 Product changes
Significant product changes that could affect safety or effectiveness require the submission of a PMA Supplement for FDA review. This applies, in particular, to unexpected side effects, device failures, or planned changes to the medical device’s manufacturing, design, or labeling.
The FDA distinguishes between different supplement types, which are based on the significance of the change and its effect on the safety of the device:
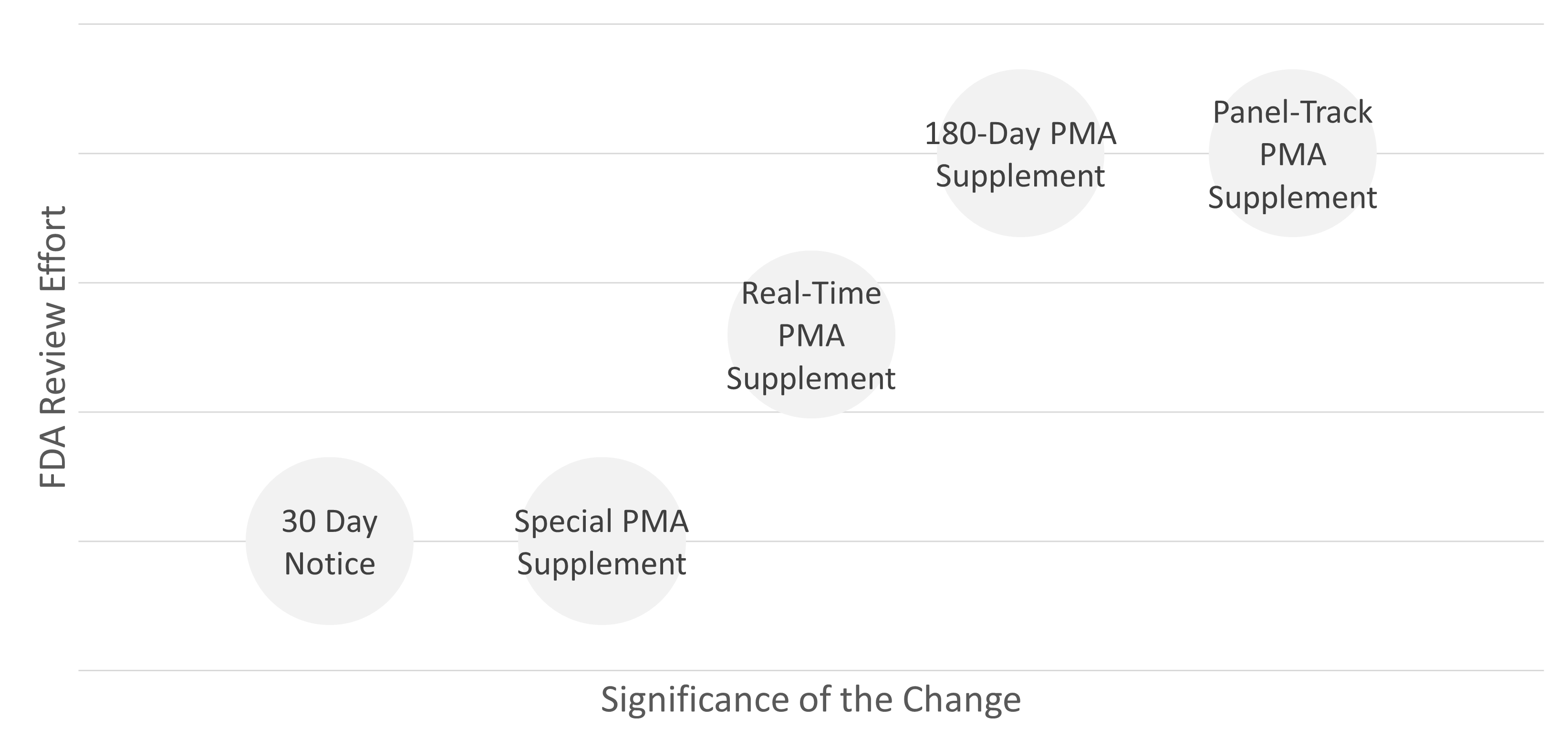
| PMA supplement type | Explanation |
| Panel-Track PMA Supplement | A change notification to an approved original PMA or Premarket Report that represents a significant change in the design or performance of the device or a new indication and for which clinical data are required to demonstrate safety and effectiveness. |
| 180-Day PMA Supplement | A change notification to an approved original PMA or Premarket Report, other than a panel track supplement, that is a significant modification to the components, materials, design, specifications, software, color additives, or labeling. |
| Real-Time PMA Supplement | A change notification to an approved original PMA or Premarket Report that represents a minor change in the device’s design, software, sterilization, or labeling. The applicant has requested an approved meeting with the FDA to review the supplement with the FDA. |
| Special PMA Supplement | A change notification to an approved original PMA or Premarket Report to improve the safety or the safer use of the device, as well as for labeling adjustments to increase product safety. These changes may be implemented by the applicant before the FDA has approved the supplement. |
| 30-Day Notice | For changes to the manufacturing process or manufacturing method that could affect the safety and efficacy of the device |
6. Conclusion and summary
The FDA’s PMA process is a complex hurdle for medical device manufacturers, but one that can be overcome.
The core elements are complete scientific documentation, comprehensive preclinical and clinical study data, and a robust quality management system. The FDA not only reviews the technical documentation but also assesses the safety and effectiveness of the medical device in all its aspects.
Approval is not a one-time event but the beginning of an ongoing regulatory responsibility. Post-approval requirements such as annual reports, safety reports, and change notifications are just as much a part of the process as FDA inspections.
For companies, this means that those who see the PMA process not as an administrative hurdle but as a strategic process for market launch will be successful.
Please feel free to contact us if you have any questions about PMA. Would you like support or an exchange on your strategic approach, answers to questions about your documents or administrative issues? Use our contact form.
The Johner Institute team is happy to help!


