The terms maintenance, preventive maintenance, restoration, inspection, service, and safety inspections are not synonymous. But they all refer to activities in the life cycle of medical devices that serve the objective of ensuring the safety, performance, and effectiveness of these devices even after they have been placed on the market.
Manufacturers and operators must meet regulatory requirements for preventive maintenance and maintenance. These specifications are often vague. Do standards such as ISO 5137 and DIN EN 62353 provide helpful assistance?
1. The terms: Preventive maintenance, maintenance, restoration
a) Definitions
Neither ISO 13485 nor the MDR define terms such as maintenance. However, they do use them.
The German MPBetreibV provides hints on how the terms are to be understood:
Maintenance of medical devices includes, in particular, servicing and repair. Maintenance measures are, in particular, inspections and servicing that are necessary to ensure the safe and proper operation of medical devices on an ongoing basis.
Translated from source § 7 MPBetreibV
Maintenance is thus the generic term that encompasses preventive maintenance.

IEC also follows this hierarchy:
combination of all technical and management actions intended to retain an item in, or restore it to, a state in which it can perform as required
Source: IEC Elektropedia
These maintenance measures include preventive maintenance:
maintenance carried out to mitigate degradation and reduce the probability of failure
Source: IEC Electropedia
Measures to delay the depletion of the existing wear stock
Source: DIN 31051:2003
Inspection is also one of the maintenance measures.
Determination of conformity with specified requirements
Source: ISO 9000:2015 3.11.7
b) Examples
When a medical technician measures a device to see if it is still performing as promised, that would be an inspection. An inspection also includes checks to see if
- consumable items, such as lubricants, need to be replaced,
- worn out items need to be replaced,
- loose cables need to be reattached, screws need to be retightened, and
- the device is dirty beyond the intended level and needs to be cleaned.
The metrological and safety inspections required by the MPBetreibV are special cases of these inspections (see Fig. 1).
The replacement of parts that are in danger of soon becoming worn or broken would be a preventive maintenance measure. Examples of such parts are hoses, rubber sleeves, and gaskets.
If defective or worn parts are replaced or repaired (e.g., glued), this is called restoration.
2. Regulatory requirements for (preventive) maintenance
a) MDR (mainly concerns the manufacturers)
The MDR does not make any precise demands on maintenance or preventive maintenance. Among other things, it requires that “devices are designed and manufactured in such a way that […] risks are eliminated or reduced as far as possible.” This explicitly includes:
risks arising where maintenance or calibration are not possible (as with implants), from ageing of materials used or loss of accuracy of any measuring or control mechanism.
MDR Annex I, Section 14.2
If maintenance is to be performed, the regulation requires:
Devices shall be designed and manufactured in such a way that adjustment, calibration, and maintenance can be done safely and effectively.
MDR Annex I, Section 14.4
For radiation emitting devices, manufacturers must describe “maintenance procedures” in the operating instructions (MDR Annex I, Section 16.1).
The instructions for use should also describe the “details of the nature, and frequency, of preventive and regular maintenance” (MDR Annex I, Section 23.4.k)).
b) MPBetreibV (concerns the operators)
The MPBetreibV requires from operators such as hospitals:
- “Maintenance measures must be carried out taking into account the manufacturer’s specifications.”
- The competence of the persons involved in these activities
- Documentation of these activities (in the medical devices book)
- Safety inspections (SI) for devices in Annex I
- Metrological inspections (MI) for devices of Annex II
The MPBetreibV not only requires the SI and MI for the respective product groups but also explicitly requires maintenance measures to be carried out in accordance with the manufacturer’s specifications.
Operators in countries other than Germany must also follow the manufacturer’s specifications. These specifications serve as risk control measures.
c) ISO 13485 (concerns mainly the manufacturers)
ISO 13485 does not generally require medical device manufacturers to maintain their devices. However, if manufacturers intend to carry out such maintenance, they must ensure the following:
- Description of the maintenance procedures
- Reference materials and reference measurements required for this purpose, if applicable
- Control of the suppliers who carry out these activities on behalf of the manufacturer
- The documentation of the activities
d) IEC 62304 and IEC 82304 (concerns mainly the manufacturers)
IEC 62304 requires a maintenance process in Chapter 6, and IEC 82304 does the same in Chapter 8.2. However, the term maintenance must be understood differently in the software context. This is because software maintenance always involves changes to the device’s design.
Nevertheless, there is a reference in these standards to maintenance/preventive maintenance in the sense of this article: IEC 82304 requires in Chapter 7.2.3.1 that the manufacturer specifies the maintenance requirements in the accompanying materials. The standard includes requirements for:
- Handling audit logs (evaluate, delete)
- Maintenance of the database
- Changing storage media
e) Further specifications for Germany
In the context of maintenance and preventive maintenance, operators should consider the following regulatory requirements:
- X-ray and Radiation Protection Ordinance (German ordinance)
- DIN EN 50678 VDE 0701:2021-02 and DIN EN 50699 VDE 0702:2021-06 for repair and re-testing of electrical equipment
- DGUV Regulation 3 entitled “Electrical installations and equipment”
f) FDA (concerns mainly the manufacturers)
The FDA specifies the requirements for “service” in more detail in 21 CFR 820.200. Thus, this law prescribes the contents of the service report (e.g., device type, device instance, date, the person performing the service, activities, and outputs).
However, the FDA will adopt the requirements of ISO 13485.
3. ISO CD 5137
The standard was in “Committee Draft” status when writing this article. Its quality is/was still inadequate, so it is not recommended for purchase. The standard can serve as a source of ideas; it does not appear suitable for implementation in its current form.
According to the ISO website, the standard is in the next status.
If you are in a hurry, skip this chapter.
a) Scope and structure
ISO 5137 is a standard specifically aimed at health institutions such as hospitals and laboratories. It comprises 29 chapters and ten annexes. A mind map with the chapter structure can be found in this document for download.
b) Essential requirements
bESP: biomedial Engineering Services Provider
The standard requires a “biomedial Engineering Services Provider” (bESP) who is employed or can act as an external service provider.
any in-house or third-party personnel, organization, responsible to carry out, implement or manage medical equipment maintenance activities
In Chapter 5 (“Responsibilities”), ISO 5137 determines the tasks of these “biomedical Engineering Services Providers” (bESP). These include the implementation of the maintenance program, the performance of annual internal audits, and the provision of the necessary resources.
The standard does not specify any concrete requirements for the competence of the bESPs. However, Chapter 6 specifies the criteria the health institution must use to determine these competencies.
Chapter overview
The table in the document linked above provides an overview of the requirements of the first 20 chapters as an example and comments on them.
c) Assessment of ISO CD 5127
It is to be hoped that the standard will not be adopted in this form. The potential for improvement is too extensive:
- The standard confuses objectives and requirements.
- The requirements seem to have been put together arbitrarily. They are partly banal and partly not applicable to many product classes.
- A consistent, process-oriented approach, even specifications for integration into a QM system, which most health institutions must have, is not apparent.
- With 30 chapters, the structure of the standard is confusing and inconsistent.
- The standard defines terms and abbreviations too late, incorrectly, or not at all. It uses terms inconsistently, even in connection with its own standard’s normative references.
- The standard contradicts internationally valid specifications with its concepts (e.g., the tags).
Thus, the standard in its current form can serve as a source of suggestions but is not yet suitable for implementation in practice.
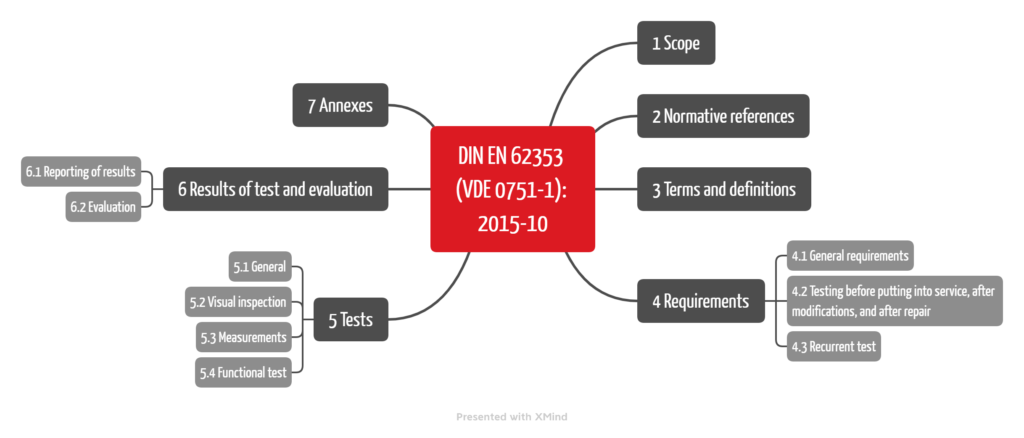
4. DIN EN 62353/VDE 0751-1
DIN EN 62353/VDE 0751-1 is entitled “Recurrent test and test after repair of medical electrical equipment.”
a) Scope
This standard is pleasantly concise and clearly structured (see Fig. 2). However, it only addresses medical electrical equipment or medical electrical systems – i.e., the devices or systems that fall within the scope of IEC 60601-1.
However, the standard can also be applied to other devices such as laboratory products.
b) Requirements
The standard “only” specifies requirements for the inspection/tests of these devices. It does not specify any requirements for maintenance, e.g., for repair, replacement, or modification of components. However, it should be used for inspections after (!) a repair.
DIN EN 62353 specifies how the tests (measurements, inspections, functional tests) are to be carried out.
At the Swedish Standards Institute you can view a comprehensive preview of an older version of the standard free of charge.
For a few euros you can purchase IEC 62353 here.
5. Tips and checklists
a) Tips for manufacturers
Development phase considerations
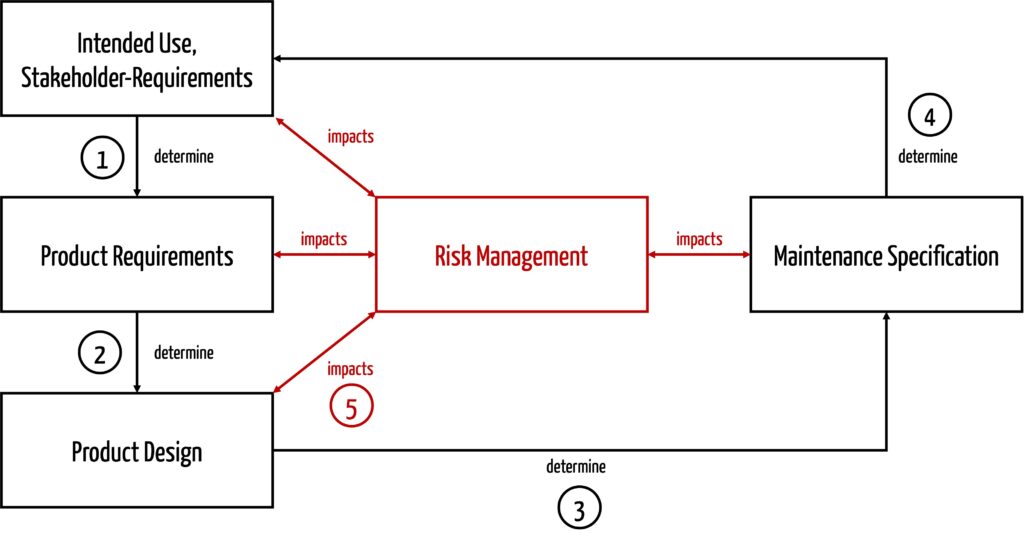
Manufacturers should consider the following influencing factors when setting maintenance or preventive maintenance specifications (see Fig. 3):
- The intended use and stakeholder requirements (e.g., lifespan, the context of use, frequency of use, and intended users) determine the product requirements. Lifespan even corresponds to a product requirement.
- Product requirements drive the design of the device. Lifespan and frequency of use, for example, must be considered by manufacturers when selecting materials and components.
- Once the device is designed, it becomes clear what actions are needed to continue to manage the risks during the use phase, in this case: what maintenance activities are needed.
- Inherently safe devices are best. These are devices that also have no inherent risks from maintenance because, for example, no maintenance is required.
- The specified maintenance activities are again part of intended use, which must be formulated accordingly.
- Risk management influences all activities. In particular, it helps to determine the necessary maintenance activities.
When defining the maintenance measures, manufacturers should pay attention to the following:
- Maintenance measures must be derived from risk management.
- The possible measures are not limited to metrological and safety inspections.
- Manufacturers must ensure the usability of the maintenance instructions (compliant with IEC 62366-1). For this purpose, they must know, define and, if necessary, create the competence of those who work with these instructions.
- The requirements of ISO 13485, which concern, for example, “suppliers” commissioned with maintenance, must be considered.
Read more about the requirements for accompanying materials such as instructions for use here.
Checklist for the post-market phase
Manufacturers must systematically collect information to ensure the safety, performance, and effectiveness of their devices throughout the product life cycle. In doing so, it is important to consider the following:
- Is the device actually being used as suspected? (User, frequency of use, use environment)
- Is contamination and wear in line with assumptions?
- Are maintenance activities performed as specified? (Frequency, competencies, materials, compliant with work instructions)
Checklist for the contents of maintenance instructions
The instructions must be complete. Typical elements of such a maintenance instruction are:
- Clear identification of the medical device to which the instruction refers
- Competence of the persons assumed to be responsible for the maintenance
- Frequency, timing, or occasions of maintenance activities
- The individual steps involved in maintenance, with photos if necessary
- Inspections (instruction, expected outputs with pass-fail criteria) to evaluate the success of the maintenance activities
- The materials, components, and tools to be used for maintenance (with the source of supply and identification, e.g., order number, if applicable)
- Specifications for documentation, if necessary, with forms or software applications
- Reporting obligations to the manufacturer and the authorities
b) Tips for operators
Checklist for purchasing devices
Purchasing should pay attention to the following points, among others:
- Does the manufacturer describe the maintenance measures precisely enough so that the company’s own team can carry them out?
- Does the manufacturer allow the operator himself or an organization commissioned by him to carry out the maintenance?
- Does the own team have the prescribed competencies?
- Does the manufacturer guarantee the supply of spare parts and consumables as well as maintenance over the complete lifetime of the device?
- Are the prices for these parts and materials, as well as for the maintenance measures by the manufacturer realistic, and are they guaranteed during the device’s lifetime?
The fact that manufacturers also want to earn money with “service” is legitimate. However, the operator should be able to obtain transparency about efforts and costs in order to make an informed selection of manufacturers and devices.
Checklist for maintenance
Operators should also consider these tips:
- Maintenance is integrated as a process in the company’s own quality management system. A separate quality assurance program, as provided by ISO 5137, creates unnecessary overhead.
- The maintenance measures provided by the manufacturer are carried out for each device, even if they do not involve measurement and safety checks. There is a work instruction for this, which defines who carries this out, when, how, and how often.
- A work instruction determines how the maintenance measures are documented in the medical device book and who checks this, how, and how often.
- It is documented for each device who may perform the maintenance (manufacturer, operator, or an organization commissioned by him).
- It is documented for each device who reports which problems to the manufacturer or the authorities.
Unfortunately, maintenance is also being misused by “maintenance/SI organizations” that sell hospitals and physicians overpriced maintenance/SI, claiming that this is required by law, even though this is not true in individual cases.
Operators depend not only on the support of their medical technology staff but also on the support of users. In this regard, a culture in which it is acceptable to use worn-out drills, equipment with brittle power cords, and 20-year-old X-ray machines is fatal.
6. FAQ on the maintenance and service of medical devices
a) Who is allowed to check medical devices?
It is the task of the manufacturers to define the requirements for the maintenance/service of medical devices. That also includes the requirements for the (competencies of the) persons who carry out these activities.
If the instructions for use accompanying the medical device explain how to carry out maintenance and service, the health institution can assume that it may carry out these activities using trained specialists such as medical technicians. However, the health institution should check with the manufacturer if these instructions are missing.
b) How often do medical devices need to be checked?
Two sources determine this frequency:
- The manufacturer’s specifications, typically in the accompanying materials such as instructions for use and
- in Germany, e.g., the Medical Devices Operator Ordinance (MPBetreibV)
The higher frequency is decisive if both sources provide information on the specific device. If only one source specifies the frequency, that one must be used. It is up to the operator if there is no information or specifications. In most cases, the restoration is carried out as and when needed.
c) What is the validation of medical devices?
The term “validation” should not be used in the context of maintenance and servicing. Validation is not(!) the check to see whether a medical device behaves according to the specifications. That would be a verification. Validation is the inspection to see whether the medical device fulfills its intended purpose as assigned by the manufacturer. A central aspect of validation is the clinical evaluation.
Please refer to the following articles on the verification and validation of medical devices and the validation of IVD.
7. Conclusion and summary
Manufacturers have a high responsibility for the safety, performance, and effectiveness of their medical devices. This responsibility concerns the complete product life cycle, including operation and maintenance.
However, the responsibility does not lie solely with the manufacturers but also with the operators and users. Legal requirements such as the MPBetreibV and standards provide a framework and guidance for action.
Unfortunately, these standards, like the colloquial language, sometimes lack clear concepts. This leads to carelessness and uncertainty. On the one hand, this results in disadvantages for patients, and on the other hand, disadvantages for operators who can be “talked into” unnecessary and expensive maintenance measures.
Maintenance must be a risk-based process as part of a quality management system for both manufacturers and operators.
The Johner Institute supports medical device manufacturers in creating precise and comprehensible maintenance instructions that comply with standards and laws and testing them for usability. Please feel free to contact us via our contact form.
Change history
- 2024-12-16: Chapter 6 (FAQ) added
- 2023-03-04: Chapter 2.c) on IEC 62304 and IEC 82304 and note added that ISO 5137 is in the next draft status
