This article describes the requirements of the in vitro diagnostic medical device regulation (IVDR) for software development and documentation.
The requirements apply to software that is part of an IVD (embedded software) and to software that is an IVD itself (“standalone” software).
This article also compares the software requirements of the MDR and the IVDR.
1. IVD > IVDR: Overview of the most important changes
The directive IVDD, which was succeeded by the IVDR, placed almost no explicit software development and documentation requirements. The IVDR has fundamentally changed this. The most critical changes in this regard are:
- The IVDR explicitly includes software in the definition of “IVD”.
- In addition, the IVDR requires software life cycle processes, including software verification and validation.
- The IVDR requires that manufacturers ensure the security of their devices.
- The IVDR places explicit requirements on software documentation.
- IVD that are software themselve (“standalone” software) must also have a unique device identification.
- There is one classification rule specifically addressing software.
You can find more information on these aspects below.
2. Definition: IVDR includes software
The definition of the term “in vitro medical device” has changed slightly compared to the IVDD:
“any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information […] “
Source: IVDR
The changes are shown in bold. In the context of software, the extended intended purposes is also interesting. It explicitly includes the “predisposition for a certain health condition or disease” and the “definition or monitoring of therapeutic measures” (e.g., medication), now.
Read more about IVD software here.
3. General safety and performance requirements of the IVDR
a) State-of-the-art software development
The IVDR has the following requirements with regard to software (identical to the MDR):
“For devices that incorporate software or for software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation.“
Most manufacturers are likely to demonstrate conformity by complying with IEC 62304. However, the standard does not address validation, and its requirements regarding IT security are also very rudimentary.
b) Reliability, “single-fault tolerance”
The IVDR explicitly extends the requirement for reliability and robustness to stand-alone software. This new requirement is very reminiscent of IEC 60601-1 and its concept of single fault condition safety:
“In the event of a single fault condition, appropriate means shall be adopted to eliminate or reduce as far as possible consequent risks or impairment of performance“.
c) Special features of mobile platforms
Just like the MDR, the IVDR specifies special requirements for mobile platforms. Manufacturers must consider their unique features:
- screen size and contrast ratios
- use environment such as physical environment (brightness, volume)
- operating systems
The wording “consider” means likely to refer to software requirements specification, risk management, and usability engineering.
d) IT security and network requirements
Without being specific, the IVDR requires manufacturers to define hardware, network characteristics, and IT security requirements. Regarding IT security, the IVDR only addresses protection against unauthorized access. Reducing IT security to this aspect is certainly questionable.
You can read more about other aspects of IT safety here. The Medical Device University provides specific examples of IT safety requirements and network characteristics.
e) Requirements for instructions for use
Manufacturers must include these minimum requirements for IT security measures, networks, and hardware in the instructions for use.
f) First overview
If you look at the requirements that the IVDR places on software, one thing stands out: except maintainability, the IVDR requires all the software quality criteria listed in ISO 25010. The only difference is that the IVDR does not do so in a structured and consistent way.
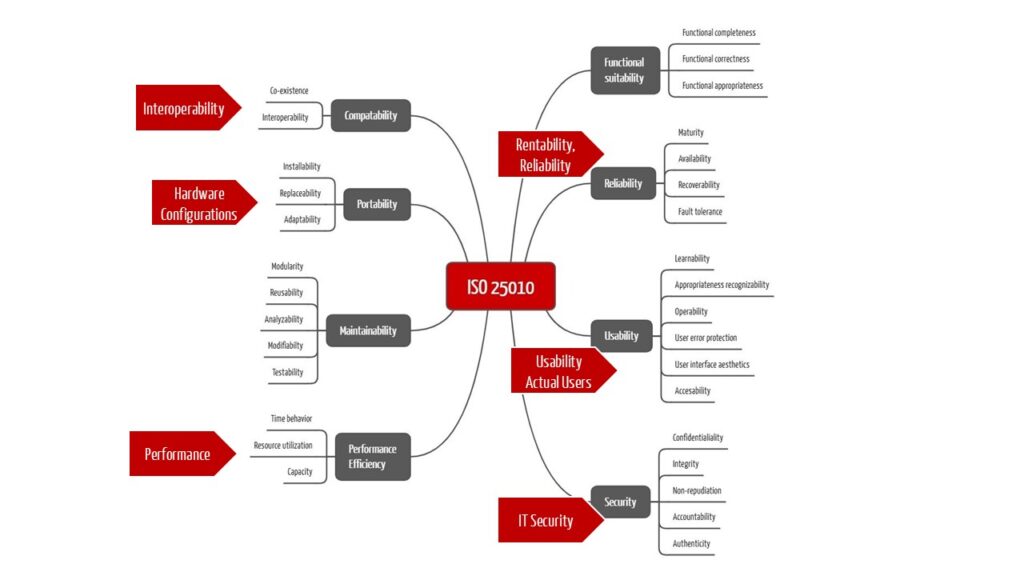
4. Software documentation in compliance with IVDR
Under the IVDD, only Annex I with the “general safety and performance requirements” was known. In contrast, the IVDR differentiates between Annex I with the “general safety and performance requirements” and Annex II with the requirements for the technical documentation.
This software documentation must include the following:
- presentation of the development stages
- description of the software and an overview of the software system (whatever is meant by this)
- description of the data evaluation, in particular, the software algorithms
- summary of the software verification and software validation. These must be carried out “in-house and in an actual user environment, ” considering various hardware configurations and possibly operating systems
A description of the software’s (at least rough) architecture and tests are likely to be mandatory. Software development, according to safety class A of IEC 62304:2006, is thus no longer sufficient to assume conformity with the IVDR’s requirements.
The requirements for tests in the “actual user environment” even go beyond those of IEC 62304. They are more aimed at usability validation with representative users in a representative user environment by IEC 62366.
5. Classification of IVD software
The manufacturers of IVD software are not required to comply with a classification rule such as rule 11 in the MDR. The only software-specific rule is:
“Software intended to control a device or affect its use is in the same class as that device. If the software is independent of any device, it is in a class of its own.”
It is consistent that the IVDR does not establish any further rules for software and thus defines the rules specifically for the analytes.
Use our innovative e-learning platform to learn how to qualify your IVD safely, how the classification of IVDs works under the IVDR, how to create a plan for implementing regulatory requirements, and finally, how to check the conformity of your file.
6. UDI
The requirements for Unique Device Identification are identical to those of the MDR.
Read more about Unique Device Identification (including for stand-alone software) here.
7. Comparison of the MDR and IVDR requirements for software development
The MDR and IVDR requirements for software development are not identical. The Johner Institute presents the most important similarities and differences in the following.
a) “General requirements“
The general requirements, known as general safety and performance requirements, are worded identically in the MDR and IVDR:
| MDR | IVDR |
| General requirements (Annex I) | |
| 14.2. Devices shall be designed and manufactured in such a way as to remove or reduce as far as possible: […] d) the risks associated with the possible negative interaction between software and the IT environment within which it operates and interacts; | 13.2. Devices shall be designed and manufactured in such a way as to remove or reduce as far as possible: […] d) the risks associated with the possible negative interaction between software and the IT environment within which it operates and interacts; |
| 17.1. Devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, shall be designed to ensure repeatability, reliability and performance in line with their intended use. In the event of a single fault condition, appropriate means shall be adopted to eliminate or reduce as far as possible consequent risks or impairment of performance. | 16.1. Devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, shall be designed to ensure repeatability, reliability and performance in line with their intended use. In the event of a single fault condition, appropriate means shall be adopted to eliminate or reduce as far as possible consequent risks or impairment of performance. |
| 17.2. For devices that incorporate software or for software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation. | 16.2. For devices that incorporate software or for software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation. |
| 17.3. Software referred to in this Section that is intended to be used in combination with mobile computing platforms shall be designed and manufactured taking into account the specific features of the mobile platform (e.g. size and contrast ratio of the screen) and the external factors related to their use (varying environment as regards level of light or noise). | 16.3. Software referred to in this Section that is intended to be used in combination with mobile computing platforms shall be designed and manufactured taking into account the specific features of the mobile platform (e.g. size and contrast ratio of the screen) and the external factors related to their use (varying environment as regards level of light or noise). |
| 17.4. Manufacturers shall set out minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended. | 16.4. Manufacturers shall set out minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended. |
| 23.4 The instructions for use shall contain all of the following particulars: […] ab) for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended. | 20.4.1 The instructions for use shall contain all of the following particulars: […] ah) for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended. |
b) Technical documentation
In contrast to the MDD and IVDD, the MDR and IVDR separate the general requirements from the requirements for technical documentation.
The requirements of the MDR and IVDR differ:
| Technical documentation (Annex II) | |
| 1.1 Device description and specification j) a general description of the key functional elements, e.g. its parts/components (including software if appropriate), its formulation, its composition, its functionality and, where relevant, its qualitative and quantitative composition. Where appropriate, this shall include labelled pictorial representations (e.g. diagrams, photographs, and drawings), clearly indicating key parts/components, including sufficient explanation to understand the drawings and diagrams; | 1.1. Device description and specification g) the description of the components and where appropriate, the description of the reactive ingredients of relevant components such as antibodies, antigens, nucleic acid primers; k) a description of any software to be used with the device; |
| 3.1. Design information Information to allow the design stages applied to the device to be understood shall include: b) for instruments, a description of major subsystems, analytical technology such as operating principles and control mechanisms, dedicated computer hardware and software; c) for instruments and software, an overview of the entire system; d) for software, a description of the data interpretation methodology, namely the algorithm; | |
| 6. Product verification and validation 6.1 Pre-clinical and clinical data […] b) detailed information regarding test design, complete test or study protocols, methods of data analysis, in addition to data summaries and test conclusions regarding in particular […] — software verification and validation (describing the software design and development process and evidence of the validation of the software, as used in the finished device. This information shall typically include the summary results of all verification, validation and testing performed both in-house and in a simulated or actual user environment prior to final release. It shall also address all of the different hardware configurations and, where applicable, operating systems identified in the information supplied by the manufacturer); | 6. Product verification and validation 6.4. Software verification and validation The documentation shall contain evidence of the validation of the software, as it is used in the finished device. Such information shall typically include the summary results of all verification, validation and testing performed in-house and applicable in an actual user environment prior to final release. It shall also address all of the different hardware configurations and, where applicable, operating systems identified in the labelling. |
Both EU regulations demand that manufacturers describe the device structure and software. The IVDR specifically mentions algorithms. Both regulations also demand testing “in-house” and “in a simulated or actual user environment” or in an “actual user environment.” They also demand testing on “different hardware configurations” and operating systems.
The MDR may have a broader understanding of the term “validation of software” than the IVDR: The MDR includes a description of the development process and the “software design.” It obviously sees validation as a synonym for constructive and analytical software quality assurance, in line with the FDA Software Validation Guidance Document. Whether these differences are really intentional can only be speculated. Both EU regulations do not differentiate by risk in their requirements on technical documentation. The regulations do not consider a safety classification, such as in IEC 62304.
This means that the requirements of the regulations for class A software go beyond the requirements of IEC 62304.
c) Unique Device Identification UDI
The specifications of the MDR and IVDR for the assignment of the UDI are identical in terms of the letters used.
d) Classification rules
The IVDR classifies the devices according to the test samples and parameters to be determined. Therefore, an analogy to “rule 11” of the MDR is not applicable.
| Classification | |
| 3.3. Software, which drives a device or influences the use of a device, shall fall within the same class as the device. If the software is independent of any other device, it shall be classified in its own right. | 1.4. Software, which drives a device or influences the use of a device, shall fall within the same class as the device. If the software is independent of any other device, it shall be classified in its own right. |
| 6.3. Rule 11 Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause: […] |
e) Clinical evaluation and performance evaluation
The requirements for clinical evaluation according to MDR and performance evaluation according to IVDR naturally differ significantly. For example, a performance evaluation does not require a discussion of equivalence (which includes software algorithms). However, it must consider the reference databases and other data sources.
| Clinical evaluation / Performance evaluation | |
| 3. A clinical evaluation may be based on clinical data relating to a device for which equivalence to the device in question can be demonstrated. The following technical, biological and clinical characteristics shall be taken into consideration for the demonstration of equivalence: — Technical: the device is of similar design; is used under similar conditions of use; has similar specifications and properties including physicochemical properties such as intensity of energy, tensile strength, viscosity, surface characteristics, wavelength and software algorithms; uses similar deployment methods, where relevant; has similar principles of operation and critical performance requirements; | |
| 1.1 As a general rule, the performance evaluation plan shall include at least: — for software qualified as a device, an identification and specification of reference databases and other sources of data used as the basis for its decision making; |
f) Conclusion and summary
While the MDR and IVDR contain identical provisions on the “general requirements” and the “UDI,” the requirements of the two EU regulations differ in the area of technical documentation (in particular, the development process and architecture). Due to the different purposes, further differences can be found in the classification and clinical evaluation or performance evaluation.
8. Conclusion
With the IVDR, the European Commission has now (finally) defined software requirements as well. However, the result is far from ideal.
- Numerous wordings are misleading, and references are unclear.
- The requirements do not adequately differentiate the risks. They are too high for non-critical devices and too low or at least too vague for critical ones.
- Due to these technical errors, the authors endangered the “harmonization capability” of IEC 62304. Deliberately?
- It is incomprehensible why the requirements for medical devices (MDR) are partially different from those for in-vitro diagnostics (IVDR).
- After 25 years, the term “validation” is clearly defined for medical devices. What does the IVDR do? It reintroduces exactly this unfortunate wording (“The documentation shall provide evidence of software validation. This information shall include […] the results of all verifications, validations […]”).
- The choice of requirements seems arbitrary. Why do the authors focus on mobile platforms? Because they have become owners of one? The authors do not address the real challenges where “guidance” would have been desirable. Not a word about bioinformatics. Not a word about artificial intelligence.
The EU Commission certainly did not provide more clarity on software with the IVDR. Perhaps the Commission should examine its own regulations, which deal with requirements for competence and quality assurance.
Read more about MDR and MDR software.



{kind=link}