EN ISO 14155:2020 is a standard that has not yet been harmonized for the MDR. It is titled “Clinical investigations of medical devices for human subjects – good clinical practice.”
It describes the state of the art for medical device manufacturers in preparing, planning, conducting, and evaluating clinical investigations. It also determines the responsibilities of the parties involved, particularly the sponsor.
This article provides an overview of ISO 14155 and tips on implementing the standard to meet the regulatory requirements for clinical investigations as quickly and easily as possible.
1. ISO 14155: An introduction
1.1 Context
The MDR has increased the requirements for clinical data which medical device manufacturers must provide to demonstrate their devices’ performance, safety, and effectiveness. Consequently, an increasing number of manufacturers are now obliged to collect this data through clinical studies, particularly clinical investigations. The MDR also imposes extensive requirements on these clinical investigations.
As the harmonized standard for the MDR in the future, ISO 14155 is intended to help manufacturers demonstrate compliance with these regulatory requirements.
1.2 Scope of the ISO 14155
The standard is applicable to clinical investigations of medical devices which are conducted to demonstrate their performance, safety, and effectiveness. Other “post-market clinical investigations” (as defined in the standard) also fall within the scope of ISO 14155. These include, for example, post-market clinical follow-up (PMCF) studies.
1.3 Objectives of the standard
The standard has several objectives:
- Protection of human subjects (safety, well-being, rights)
- Reliability of results
- Clarity regarding responsibilities, in particular, those of the sponsor (e.g., manufacturer), principal investigator, and other potential participants (e.g., CRO, hospital)
- Uniform understanding among all parties involved (sponsors, investigators, ethics committees, authorities, notified bodies)

1.4 Benefits for manufacturers
Here is why medical device manufacturers benefit from the standard:
- Less regulatory uncertainty and discussion with authorities and notified bodies
- This means: faster and less costly “approval” of devices
- Time savings thanks to specification of requirements and examples, e.g., for the structure of documents such as the clinical evaluation plan and the clinical evaluation report
2. Structure of ISO 14155:2020
The standard comprises over 90 pages and consists of ten chapters and ten annexes (see Fig. 2).
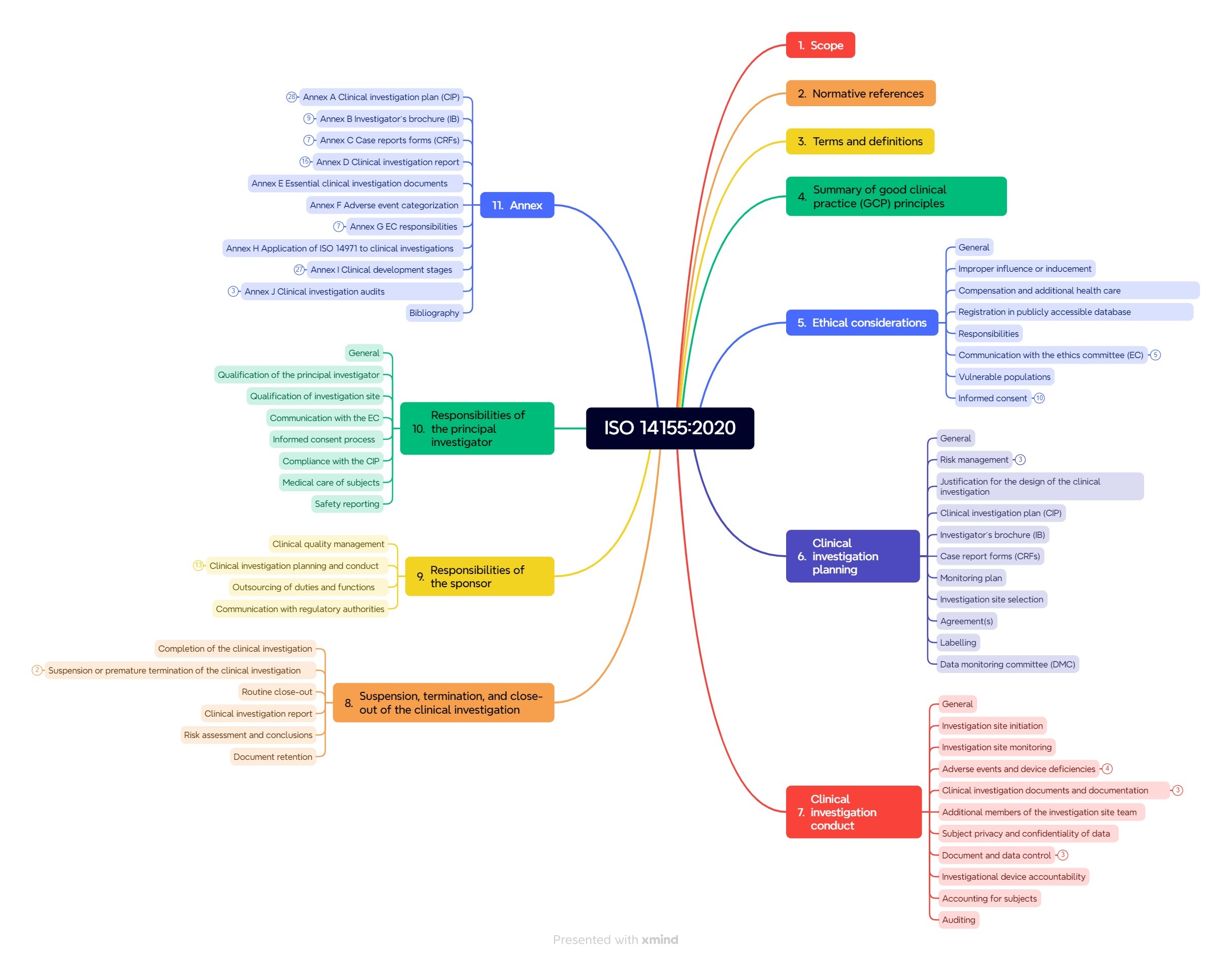
Chapter 4: Good clinical practice
Chapter 4 summarizes the principles of good clinical practice (GCP).
Chapter 5: Ethical considerations
Chapter 5 contains ethical considerations. It addresses, among other things, communication with the ethics committee, responsibilities, vulnerable groups, informed consent, and patient information.
Chapter 6: Clinical investigation planning
Chapter 6 describes the planning of the clinical investigation. It includes:
- Requirements for the investigational device
- Requirements for the design of the clinical investigation
- Requirements for the selection of the trial site and the essential documents of the clinical investigation
These documents include:
- Clinical Investigation Plan (CIP)
- Investigators’ Brochure (IB)
- Case Report Forms (CRFs)
The annexes provide further information on the structure of these documents.
Chapter 7: Clinical investigation conduct
Chapter 7 specifies the requirements for conducting clinical investigations:
- Information on practical implementation
- Reporting of events or device defects
- Handling of documents and data
- Conducting monitoring
- Confidentiality of data and identification of subjects
Chapter 8: Suspension, termination, and close-out of the clinical investigation
This section contains the requirements for the suspension, termination, and close-out of clinical investigations. These also apply to the clinical investigation report, risk evaluation, and document retention.
Chapter 9 and 10: Responsibilities
Chapters 9 and 10 regulate the responsibilities of the sponsor and the principal investigator. The requirements for the sponsor are particularly relevant for medical device manufacturers.
Normative and informative annexes
The extensive annexes to ISO 14155 are partly normative.
- Clinical investigation plan (CIP) (normative): Specified contents of the CIP
- Investigator’s brochure (IB) (normative): Minimum requirements for the contents of the IB
- Case report forms (CRFs) (informative): Tips for creating CRFs
- Clinical investigation report (normative): Specified contents of the clinical investigation report at the end of the clinical investigation
- Essential clinical investigation documents (informative): Documents that must be available at the sponsor’s or the investigation site before, during, and after the clinical investigation
- Adverse event categorization (informative): Overview table and graphics for orientation
- Responsibilities of the ethics committee (EC) (informative): Guidelines for good practice within ethics committees
- Application of ISO 14971 to clinical investigations (informative): Graphic on dealing with risks in a clinical investigation
- Clinical development stages (informative): Overview of the different types of clinical investigations that accompany the development stages of medical devices and applicability of ISO 14155 to the different types
- Clinical investigation audits (informative): Includes tips on how the sponsor can demonstrate that the clinical investigation is conducted in accordance with GCP
3. Important requirements of the ISO 14155
3.1 Ethical considerations
Clinical investigations must be conducted in accordance with ethical principles. ISO 14155 must always be applied in conjunction with the current version of the Declaration of Helsinki, which sets out the ethical principles for conducting clinical investigations.
Foreseeable risks must be weighed against the expected benefits.
The rights, safety, and well-being of the participants must take precedence over scientific interests.
3.2 Unconditional scientific rigor
The investigation must follow clear and comprehensible statistical guidelines that provide well-founded results with a high level of evidence.
Preclinical and clinical information must adequately support the investigation.
3.3 Comprehensive documentation
Thorough and comprehensible documentation of all phases of the clinical investigation is required.
The investigation must be conducted in accordance with the documents approved by the ethics committee and the authorities (including the investigation plan, investigator’s brochure, and patient information).
3.4 Clear responsibilities
Clear definitions of the roles and responsibilities of all parties involved are essential and must be specified in writing.
4. Avoidable pitfalls
4.1 Requirements not met by the manufacturer
It is often assumed that the manufacturer can easily be the sponsor in clinical investigations.
Sponsorship in a clinical investigation is subject to several requirements, including integrating clinical quality management into the manufacturer’s existing QMS.
Therefore, it is important to note that the investigation can only be initiated if you, as the sponsor, fulfill the requirements of the standard and the MDR.
4.2 Incorrect assumptions regarding the applicability of ISO 14155
“This is a PMCF study; the device is certified; the standard does not apply.”
This is an inaccurate assumption.
The standard is not only applicable to “approval studies” conducted to obtain CE certification but also, in part, to studies conducted to clarify scientific questions (Article 82 MDR) or to PMCF studies (Article 74 Studies). This is further explained in Annex I.7 of the standard.
4.3 Insufficient documentation
A summary of the study results as only documentation of the clinical investigation, is generally insufficient to prove clinical evidence.
Manufacturers must evaluate the clinical data of their own device gathered from published clinical investigations as part of the clinical evaluation. If you conduct a study with your own device, you are expected to submit the study protocol and demonstrate that the study was conducted in accordance with ISO 14155 in order to provide sufficient clinical evidence. The notified bodies check this as part of the “clinical evaluation assessment” (Section E of MDCG 2020-13).
In the worst case, doubts could arise about the quality of the data, since you as a manufacturer cannot prove that the study was conducted in accordance with ISO 14155, for example, without the study protocol and other documents. As a consequence, you might have to exclude the data.
4.4 Exclusive focus on ISO 14155
It is a common misconception that complying with ISO 14155 alone fulfills all national requirements. However, national requirements must also be taken into account, particularly outside the EU.
5. Versions of ISO 14155
5.1 Differences between versions 2012 and 2020
Several notable changes have been implemented in ISO 14155:2020-01 (or DIN EN ISO 14155:2021-05) compared to the 2012 predecessor standard:
- The link with the ICH-GCP Guideline has been emphasized more strongly, and a summary of the GCP principles has been integrated into section 4 of the standard.
- Risk management has been integrated into the entire clinical investigation process.
- Annex I, which is of particular interest to manufacturers, has been introduced to explain the applicability of the standard to the different clinical development stages of the medical device.
- A requirement to register the clinical investigation in a publicly accessible database has been added (new in version 2020/2021).
- Clinical quality management (section 9.1) and risk-based monitoring (section 6.7) have been integrated.
- The current version considers the adaptation to international data protection requirements.
5.2 Outlook for the next version
An amendment to ISO 14155 is being worked on. The publication date of the final standard is currently unknown (June 2025).
The revision aims to harmonize clinical investigations with the EU requirements of the MDR. It is to become valid without a transition period. (Source)
6. Conclusion and summary
ISO 14155 is the gold standard for the preparation, planning, conduct, and evaluation of clinical investigations of medical devices, regardless of its harmonization. It is important to note that this is not only applicable to clinical investigations; it should also be followed for studies outside Europe.
The standard is sufficiently precise and complete to represent a benefit rather than an additional regulatory burden for manufacturers.
The Johner Institute’s clinical experts help you to find clinical data and decide whether a clinical investigation is necessary.
If so, we will support you throughout your clinical investigation or PMCF study, from planning and application to implementation.
- Study design: We help with design and coordination
- Statistical sample size planning: Optimal planning for valid results
- CRO selection: We find the right contract research organization (CRO)
- Submission to authorities: Preparation of regulatory-compliant documentation
- Implementation and evaluation: Always at your side during collaboration with the CRO in the clinical field
Here is where you will find an overview of the support offered by the Johner Institute for clinical evaluations and investigations.


