GLP (Good Laboratory Practice) defines requirements for a quality assurance system for non-clinical health and environmental safety tests. It also describes the organizational procedure and conditions under which laboratory tests are planned, carried out, and monitored. GLP also covers the record and reporting of.
In this article, you can read which requirements medical device manufacturers may also have to meet, which regulations must be observed, and which of these requirements affect software.
GLP – Good Laboratory Practice: What is it?
Objectives of Good Laboratory Practice
GLP is intended to ensure the quality of the output of non-clinical laboratory studies. This applies in particular to laboratory studies that are intended to demonstrate the safety of the following product categories:
- Medicinal products
- Medical devices
- Food additives
- Plant protection products
- Cosmetic products
- Electronic devices
This list differs slightly in different jurisdictions (e.g., EU, USA/FDA).
The tests do not (only) concern the devices themselves but, above all, the “test items” contained in these devices.
Examples of laboratory tests
Examples of such tests, which are not carried out on humans, are
- Tests for the determination of physico-chemical properties, contents, concentrations
- Tests for the determination of toxicological properties (in vitro and in vivo)
- Tests for the determination of mutagenic properties (in vitro and in vivo)
- Ecotoxicological tests (effects on aquatic and terrestrial organisms and natural ecosystems, behavior in soil, water, and air)
- Tests for the determination of residues
- Analytical tests of biological materials
Differentiation from GMP (Good Manufacturing Practice) and GCP (Good Clinical Practice)
The requirements of GLP do not apply to clinical studies on humans or animals. These clinical studies are subject to the GCP requirements, among others.

Good Laboratory Practice also has nothing to do with the device’s manufacture. The GMP guidelines are to be applied here.
Requirements of Good Laboratory Practice GLP
GLP describes a quality assurance system that includes the usual requirements for quality management systems, such as management responsibility and document control, as well as the specifics of laboratory practice.
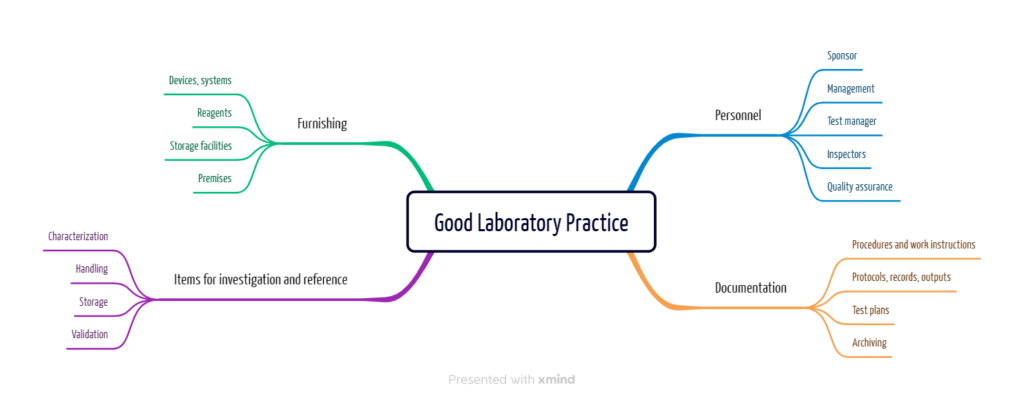
Regulatory requirements for GLP
a) To whom GLP applies
The regulatory requirements are not limited to the pharmaceutical industry, medical laboratories, or medical device manufacturers. All companies that manufacture or place the above device categories or “test objects” on the market are subject to these regulatory requirements.
Medical device manufacturers can be affected in two ways:
- Own devices
Their own devices must meet the basic requirements of the medical device directives. Tests of their properties and their safety for human health may fall under GLP. - Devices for laboratories and testing facilities
IVD medical device manufacturers, in particular, supply their devices to testing facilities. GLP also places requirements on equipment, such as the validation of computerized systems.
b) Overview of the regulations
The essential Good Laboratory Practice GLP requirements are formulated in the OECD consensus documents. Many national and international regulations have adopted their requirements.
USA
In the USA, 21 CFR part 58 (“Good Laboratory Practice for Nonclinical Laboratory Studies”) should be mentioned.
The FDA has published a comparison of 21 CFR part 58 and the OECD requirements on its website.
Europe
At the European level, two directives define the requirements for GLP:
- Directive 2004/9/EC requires EU states to determine national authorities that carry out GLP inspections. These inspections must follow the OECD guidelines “Compliance Monitoring Procedures for GLP” and “Conduct of Test Facility Inspections and Study Audits.” This directive replaces Directive 88/320/EEC.
- Directive 2004/10/EC obliges EU countries to take all measures to ensure that certified laboratories carry out tests in accordance with the OECD guidelines. This directive replaces Directive 87/17/EEC.
European nations (e.g., Germany)
The national states have transposed these European directives into national laws, such as the Chemicals Act and the GLP Administrative Regulation (ChemVwV-GLP) in the case of Germany.
In Germany, the Federal Institute for Risk Evaluation, more precisely “the GLP Federal Office at the BfR is responsible for the coordination and harmonization of GLP-relevant issues in the national and international area as well as in the monitoring of certain GLP test facilities in Germany and abroad based on the “General Administrative Regulation GLP (ChemVwV-GLP)”.”
The Chemicals Act specifies the legal requirements for Good Laboratory Practice in Germany. The requirements therefore relate to substances and substance mixtures and not specifically to medical devices.
Standards
ISO 15189 and ISO/IEC 17025 formulate requirements specifically for medical(!) laboratories and test laboratories. Laboratories are often accredited according to both standards.
Particular requirements of GLP for software
The regulatory requirements for Good Laboratory Practice GLP also apply to the use of software and computerized systems.
a) USA/FDA
As soon as computerized systems replace paper-based documents and records, the requirements of 21 CFR part 11 must be met in the USA.
Read a detailed article on 21 CFR part 11 (German).
However, the scope of the FDA Software Validation Guidance Documents refers to the development and manufacture of medical devices.
b) OECD on computerized systems
The OECD has published a consensus document, “The Application of the GLP Principles to Computerized Systems“. The requirements range from bland to comprehensible to absurd (“The source code of the application software in the test facility must be available or retrievable by the test facility”).
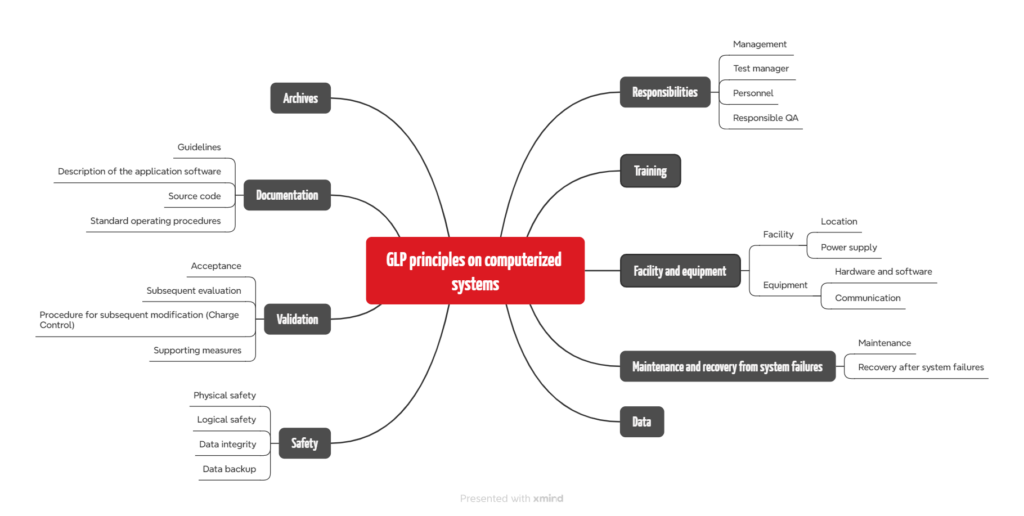
The required standard operating procedures are also of interest to medical device manufacturers:
- Operation of the computerized system (hardware/software)
- Security measures to detect and prevent unauthorized access and unauthorized program changes
- Modification of programs and their documentation, system changes (hardware/software), including any necessary tests before putting into service again
- Periodic review of the correct functioning of the entire system or individual components and their documentation
- Maintenance procedures for computerized systems and associated equipment
- Software development and instructions for carrying out acceptance tests and their documentation
- Back-up for stored data
- Disaster recovery: contingency plans for continuing tests in the event of system failures
c) EU directives
The EU directives contain only a few statements on the handling of software and computer systems, none of which go beyond the OECD requirements:
2004/9/EC states: The inspector should check […]
- that records have been kept of operation, maintenance, verification, calibration, and validation of measuring equipment and apparatus (including computerized systems),
- in automated systems, data generated as graphs, recorder traces, or computer print-outs are documented as raw data and archived,
- use, maintenance, cleaning, calibration, and validation of measuring apparatus, computerized systems and environmental control equipment;
- any changes in the raw data, including data stored in computers, did not obscure previous entries, included the reason for the change and identified the person responsible for the change and the date it was made,
- computer-generated or stored data have been identified and that the procedures to protect them against unauthorized amendments or loss are adequate,
- the computerized systems used within the study are reliable, accurate and have been validated,
- records have been kept of operation, maintenance, verification, calibration, and validation of measuring equipment and apparatus (including computerized systems),
According to 2004/10/EC, the laboratory must, among other things, […]
- implement procedures to ensure that computerized systems are suitable for their intended use and are validated, operated, and maintained in accordance with these principles of Good Laboratory Practice.
- ensure that computerized systems used in the course of an test are validated;
- equipment, including validated computerized systems, used to obtain, record, and reproduce data and control the environmental conditions relevant to the test are suitably located, designed, and sufficiently powerful.
The requirements do not go further than those of 2004/9/EC.
d) National regulations
The national regulations adopt these requirements.
In Switzerland, the Information Technology Working Group at the Federal Office of Public Health has developed guidelines for the validation of computerized systems in the GLP context.
Read more about the validation of computerized systems.
Conclusion and summary
GLP (Good Laboratory Practice) defines requirements for the QM system of laboratory facilities that carry out non-clinical safety testing of devices or substances. These devices are not limited to medical devices.
The OECD has formulated these requirements in several consensus documents. The EU, the European nation-states, and the FDA are strongly oriented toward them and, in some cases, adopt the requirements verbatim.
IVD medical device manufacturers, in particular, can use the GLP regulations to offer their customers services to achieve “GLP compliance.”
Change history
- 2024-01-11: Review up-to-dateness + formal changes/additions
- 2016-12-20: Initial creation of the article
