Most medical device manufacturers associate the term “design validation” with the FDA. However, not only the FDA but also European regulations, particularly ISO 13485, require design and development validation. Nevertheless, design validation and validation of the design should not be confused.
This article shows how the two differ and which regulatory requirements must be observed.
1. Definition des Begriffs “Design Validation”
Die FDA defines the term design validation:
„establishing by objective evidence that device specifications conform with user needs and intended use(s)“
In other words, design validation is the review to determine whether the specifications that result from the development meet the intended purpose and the user’s requirements.
Unfortunately, the FDA does not define the term “user needs“. However, ISO/IEC 25060:2014 defines the term “requirement” (user need).
a necessary prerequisite identified for a user or group of users to achieve a desired work outcome within a specific context of use
This means that the validation must check whether the users can achieve the intended purpose with the product and whether the product fulfills the stakeholder requirements resulting from the user needs, such as the technical requirements (= requirements for the work result).
2. Design validation: What is required by the regulatory authorities
a) Requirements by the FDA
The FDA requires about design validation:
Each manufacturer shall establish and maintain procedures for validating the device design. Design validation shall be performed under defined operating conditions on initial production units, lots, or batches, or their equivalents. Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate. The results of the design validation, including identification of the design, method(s), the date, and the individual(s) performing the validation, shall be documented in the DHF
21 CFR part 820.30 (g)
In this section, the FDA specifies what needs to be done for design validation, other than testing by definition and as described above, to ensure that the specifications that result from development meet the intended purpose and the requirements of the users:
- Manufacturers must assess that the medical devices (not just their specifications) comply with the intended purpose and requirements.
- This assessment must be carried out in the defined context of use.
- Products that originate from production or correspond to such products must be used for the review.
- The design validation must include software validation and risk management.
Conclusion: Design validation is the “normal” validation of a medical device or a product model (and not just a review of specifications). For non-standalone software, a final validation would also consider production aspects that are not the subject of design validation.
With the amendment of 21 CFR part 820, the FDA adopts the requirements of ISO 13485 Chapter 7.3.7 concerning design validation.
b) Requirements of ISO 13485
The ISO standard requires in Chapter 7.3.7 “Design and Development Validation” (in German: Design- und Entwicklungsvalidierung”) that the resulting product must comply with the intended purpose. In contrast to the FDA, ISO 13485 requires a clinical evaluation as part of the (design) validation. The standard points out that:
- clinical data is a result of design validation,
- manufacturers must carry out this validation with a representative product,
- before the release of the product, and
- in the intended use environment (e.g., in conjunction with other products).
3. Design validation: Avoid misunderstandings
The term “design” is often used to describe a product’s architecture in the context of its development. Each of the development steps and, thus, the “design” of the architecture should be reviewed. This review is sometimes referred to as “validation,” and therefore, the review of the architecture is called “design validation.” However, this must not be confused with design validation in the sense of ISO 13485, i.e., product validation.
Both IEC 62304 and IEC 60601-1 correctly classify the review of the architecture as “verification”.
For standalone software, design validation corresponds to the validation of the entire product since there are hardly any production aspects to consider.
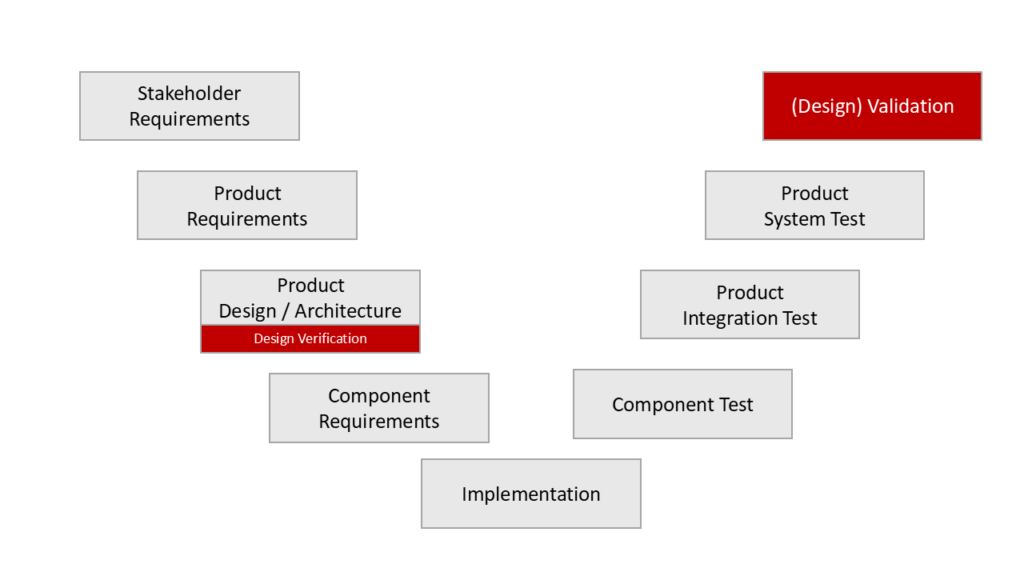
In other words, in the case of a software product, design validation is not the “validation of the software design”. Design validation corresponds to the validation of the entire software, including clinical evaluation and usability validation.
Read more about the validation of medical devices and the confusion surrounding the term software validation here.
4. Conclusion and summary
The term “validation” has multiple meanings, as does the term “design.” Therefore, it is not surprising that design validation is not always understood in the same way.
If ISO 13485 and the FDA had both spoken of product validation instead of design validation, what is meant would have been clearer. At least both regulatory requirements agree and will even be merged with the amendment of 21 CFR part 820.
Change history:
- 2024-11-04: Article largely revised and restructured; graphic replaced
- 2015-10-11: First version of the article published


