Combination products consist of the combination of a medical device and a medicinal product. Since both medical device and medicinal product law could be applicable here, there are some special features that must be taken into account for products of this type.
In this article, you learn more about
- what a combination product is,
- which law is applicable, and
- which special requirements apply to combination products.
1. What are combination products?
a) Definition under EU law
EU law does not officially include the term “combination product”. It is outlined in several regulations, but never expressly used.
Regulation EG 1394/2007 (medicinal products for novel therapies), for example, defines the term “combined advanced therapy medicinal product” as an
advanced therapy medicinal product that incorporates, as an integral part of the product, one or more medical devices.[…].
The MDR and Directive 2001/83/EC for medicinal products do not use the term “combination product” or a similar term. The MDR does, however, set out specifications for products that are a combination of a medicinal product or active substance and a medical device.
Article 1(8) and (9) of the MDR sets out rules for when manufacturers either need to use the MDR or Directive 2001/83/EC for medicinal products.
(8) Any device which, when placed on the market or put into service, incorporates, as an integral part, a substance which, if used separately, would be considered to be a medicinal product as defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma as defined in point 10 of Article 1 of that Directive, and that has an action ancillary to that of the device, shall be assessed and authorised in accordance with this Regulation.
Art. 1 Para. 8 MDR
However, if the action of that substance is principal and not ancillary to that of the device, the integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004 of the European Parliament and of the Council, as applicable. In that case, the relevant general safety and performance requirements set out in Annex I to this Regulation shall apply as far as the safety and performance of the device part are concerned.
(9) Any device which is intended to administer a medicinal product as defined in point 2 of Article 1 of Directive 2001/83/EC shall be governed by this Regulation, without prejudice to the provisions of that Directive and of Regulation (EC) No 726/2004 with regard to the medicinal product.
Art. 1 Para. 9 MDR
However, if the device intended to administer a medicinal product and the medicinal product are placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004, as applicable. In that case, the relevant general safety and performance requirements set out in Annex I to this Regulation shall apply as far as the safety and performance of the device part of the single integral product are concerned.
This enables us to conclude that the MDR is aware of the principle of “combination products”, even though it does not define them. The MDR regulations also mean that either the MDR or Directive 2001/83/EC is primarily applicable for medicinal products in the case of these products, but not both at the same time.
Interim result
There is no specific regulation, directive or law for combined products consisting of a medicinal product or active substance and a medical device. However, the rules mentioned allow us to derive the following definition of combination products:
A product is considered to be a combination product if a combination of a medicinal product or active substance and a medical device combine to form a single product.
b) Definition under US law
In contrast to the EU, the term combination product does officially exist in the US (21 CFR 3.2(e)). This term describes products that meet one of the following conditions:
Combination of medicinal products and medical devices and/or a biological product
- manufactured as one unit (physically, chemically, or otherwise), or
- co-packaged, or
- packaged separately, but both products can only achieve the intended effect together, or
- combinations intended for clinical investigations that are packaged separately and that need to be used together for the investigation.
Examples of FDA conditions (allocated to the conditions):
- Self-injector containing adrenaline or an insulin pen (1)
- Surgical sets containing instruments, covers and antimicrobial swabs (2)
- Cardioplegia set for a heart-lung machine (tubes co-packaged with the cardioplegia solution) (2)
- Product for photoimmunotherapy together with the active substance (3)
Accordingly, combination products in the sense of the FDA are:
combinations of medical devices and medicinal products that are either physically combined, co-packaged, or cross-labeled.
It is clear that the FDA understands the term “combination products” much more deeply than the MDR. Of all of the variants of “combination products,” only one overlaps with the MDR’s understanding of combination products, namely variant one, “manufactured as a unit (physically chemically, or otherwise).”
2. What are combination products NOT?
The term “combination” needs to be understood in the sense of “integrated”, “belonging to”, or “inseparable”. Combination products (medicinal product-device units) are therefore not:
a) Products for releasing medicinal products
Products that are only intended to release medicinal product (substances) and have not been tailored to a specific medicinal product are handled as medical devices (see MDR Article 1(9)1)). Examples of products of this type are syringe pumps for general purposes.
b) Systems and procedure packs
In the EU (unlike in the US), systems and procedure packs are not considered to be combination products. There are special rules for systems and procedure packs in Article 22 MDR.
“Procedure pack” means a combination of products packaged together and placed on the market with the purpose of being used for a specific medical purpose;
Source: Article 2 (10) MDR
“System” means a combination of products, either packaged together or not, which are intended to be inter-connected or combined to achieve a specific medical purpose;
Source: Article 2 (11) MDR
If systems or procedure packs of this type contain medicinal products, the provisions in Article 22 MDR (“systems and procedure packs”) apply to this aggregated entity.
If systems and procedure packs are co-packaged or cross-labeled with medicinal products, the FDA considers them to be combination products.
You can find out more about systems and procedure packs in our article on the topic.
3. Applicable law: Medical device law or medicinal product law?
Either Directive 2001/83/EC for medicinal products and Regulation 726/2004 or the MDR for medical devices applies. They are therefore either primarily treated as medicinal products or primarily as medical devices, but not as both at the same time.
Depending on the field of law that is primarily relevant, manufacturers also need to check the combination product broadly as a medicinal product or broadly as a medical device.
This has an effect, for example, on:
- which route the manufacturer needs to take to provide a certificate of conformity (medical device) or approval (medicinal product),
- the party to whom the necessary documents need to be submitted (notified body for medical devices, medicinal product authorities for medicinal products).
a) Rules of Art. 1 (8) and (9) MDR
The rules of Art. 1 (8) and (9) MDR concerning the field of law that is primarily relevant:
- All devices incorporating, as an integral part, a medicinal product that has an action ancillary to that of the device, are medical devices. However, if the main effect of the product lies with the medicinal product, it is treated as a medicinal product (MDR, Article 1 (8)).
- All medical devices intended to administer a medicinal product are medical devices, unless they are placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable. Then the product is treated as a medicinal product (MDR, Article 1 (9)).
The main ingredient is the ingredient which achieves the intended use, indication or effect.
The component is integral if both components are firmly and inseparably connected with each other. Furthermore, it is required that the combination serves a specific purpose.
Combined products must always meet the requirements from the respective Annex I of both regulations. In the MDR, these are the general safety and performance requirements; in Directive 2001/83/EC, these are analytical, toxicological, pharmacological, and medical or clinical requirements and evidence of testing of medicinal products.
Possible variants and applicable law in detail
Variants Art. 1(8) MDR
Medical device, if:
- medicinal product integral part of medical product
- supporting function medicinal product
- example: Heparin-impregnated catheter (drug has supportive effect)
Medicinal product if:
- drug integral part of the medical product
- main function drug
- example: Antiseptic patches where the main effect is the antiseptic and the patch stores the antiseptic
Variant Art. 1(9) MDR
Medical device if:
- medical product intended to deliver medicinal product according to Directive 2001/83/EC
- example: Medication pumps for refilling
Medicinal product, if:
- medical product intended to deliver medicinal products according to Directive 2001/83/EC
- inseparable total product
- medical product and medicinal product intended for use in combination only
- combination not reusable
- example: Epi-Pen (syringe pre-filled with drug) for single use
In the case of any doubt, the Directive for medicinal products applies
There are situations in which both definitions, medicinal product and medical device, apply.
Example: The primary effect of a plaster wetted with antiseptic can be claimed by the manufacturer as:
- the physical protection of the wound, and/or
- the antiseptic effect to avoid an infection could be the focus.
In domestic use the physical protection is the main feature while the antiseptic effect is more important in clinical use due to the higher germ load. The plaster is authorized for both areas.
In cases where there is doubt, the directive for medicinal products applies (see Article 2(2) 2001/83/EC and Medicinal Products Act Section 2(2) No. 7).
b) Taking into account both areas of law in the conformity assessment procedure/approval
Representatives of both areas, medicinal product law and medical device law, are involved in the conformity assessment procedure or approval.
Medical device has priority
If primarily the medical device regulation is applicable, the notified body must involve the competent authority of a member state or the EMA and obtain a scientific opinion.
Section 5.2 of Annex IX of the MDR states that in the case of a medical device containing a medicinal product as defined in Art. 1 No. 2 of Directive 2001/83/EC for medicinal products, the notified body must consult the competent authority for medicinal products. Without their positive vote, the combination product will not receive a certificate of conformity.
This is a deviation from the MDD/AIMDD. The MDR explicitly states that the notified body may not issue a certificate if the opinion of the medicinal products authority is negative (Annex IX 5.2 (e) and (f)).
Medicinal product has priority
The same applies in reverse for combination products authorized according to medicinal product law. A notified body is involved here (Article 117 MDR).
Article 117 MDR regulates the procedure for how the notified body is included in the case of combination products that are primarily medicinal products.
- If the medicinal product manufacturer uses a medical device with an EU declaration of conformity from the manufacturer, the further involvement of a notified body is not necessary. The product then already complies with Annex I MDR. The medicinal product manufacturer must submit evidence along with the application for authorization.
- If no certificate is available and the medical device needs to involve a notified body for itself for the certificate of conformity, the medicinal product manufacturer must request a “notified body opinion” from a notified body that has been appointed for products of this type. This is also about proving that the medical device part meets the requirements in Annex I of the MDR.
The rules for Companion Diagnostics (CDx) are similar. There is more about this in our article on the topic.
4. Regulatory requirements of combination products
The combination of medical device and medicinal product results in new hazards in terms of interoperability and compatibility that are also addressed in Annex I of the MDR. Other requirements apply with regard to these hazards.
a) Risk assessment
The manufacturer must demonstrate that they meet Paragraph 12 of Annex I, Article II MDR.
Annex I Paragraph 12.2 MDR:
Devices that are composed of substances or of combinations of substances that are intended to be introduced into the human body, and that are absorbed by or locally dispersed in the human body shall comply, where applicable and in a manner limited to the aspects not covered by this Regulation, with the relevant requirements laid down in Annex I to Directive 2001/83/EC for the evaluation of absorption, distribution, metabolism, excretion, local tolerance, toxicity, interaction with other devices, medicinal products or other substances and potential for adverse reactions, as required by the applicable conformity assessment procedure under this Regulation.
The impact assessment also applies to certain manufacturing technologies, such as sterilization (Annex I Paragraph 11.4); the manufacturer should evaluate whether the technology/method used has any effect on the safety and performance of the medical device part. In addition to this, the authority responsible for evaluating the approval dossier (EMA) and/or (NCAs) focuses on the evaluation of medicinal product part.
b) Documentation
Annex II Paragraph 6.2 a) MDR states:
Where a device incorporates, as an integral part, a substance which, if used separately, may be considered to be a medicinal product within the meaning of point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma, as referred to in the first subparagraph of Article 1(8), a statement indicating this fact. In this case, the documentation shall identify the source of that substance and contain the data of the tests conducted to assess its safety, quality and usefulness, taking account of the intended purpose of the device.
Information must therefore be provided on the medical device on:
- the presence of a medicinal product
- the source of the medicinal product
- the data of the tests conducted to prove its safety, quality and usefulness.
c) Usability
For individual product classes, the FDA has published special guidance documents on human factors engineering. These include a guidance document for combination products.
d) FDA
The FDA describes its requirements for combination products, at least indirectly, in its Guidance Document “How to Prepare a Pre-Request for Designation”.
5. Classification
a) Classification rules
The conformity assessment procedure and the approval (as described in medicinal product law) are initially based on whether the product needs to be qualified as a medicinal product or a medical device.
If the MDR primarily applies, the combination products come under risk class III according to Rule 14.
All devices incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product, as defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive, and that has an action ancillary to that of the devices, are classified as class III.
7.1. Rule 14
High requirements are placed on class III products according to the MDR. This means a high cost
- in terms of documentation
- in terms of monitoring
- less choice of notified body
b) Regulatory strategy appropriate for the classification (examples)
Manufacturers should be strategic in order to get a combination product through the conformity assessment procedure or the approval procedure as well as possible. They may choose, for example, the following strategies:
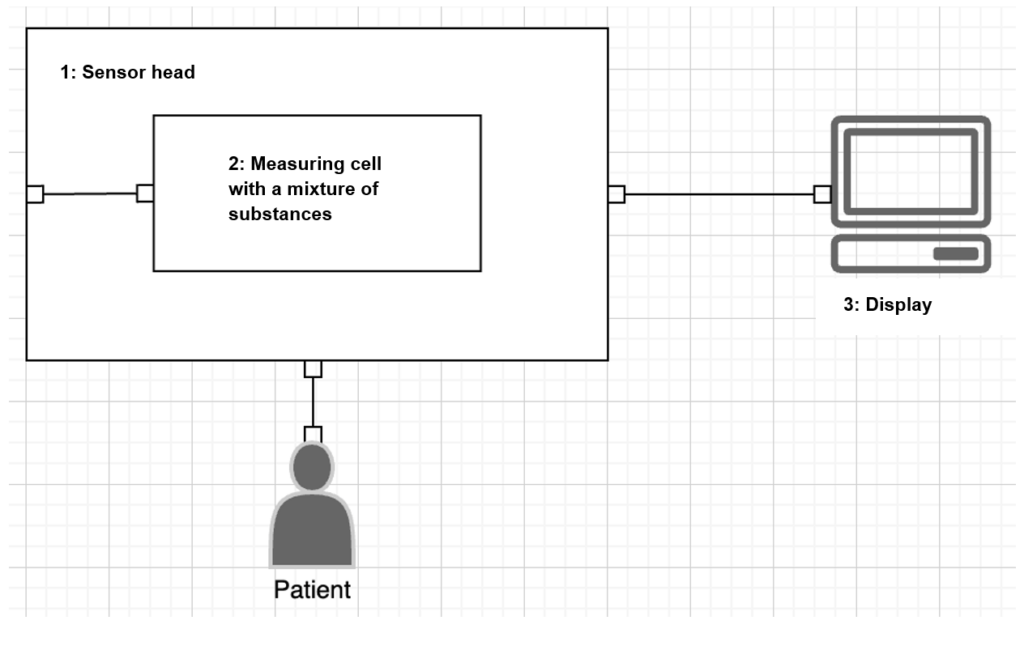
The purpose of the product is to measure an enzyme level via the skin. The sensor consists of the sensor head (1) with a measuring chamber and a measuring cell (2) with a mixture of substances that generates the measuring signal in the exchange with tissue fluid. The measuring signal is calculated into a measurement value and shown on the display (3).
Regulatory strategy
| Combined product parts | The mixture of substances 92) is used to make a medical diagnosis and must be qualified as a medicinal product according to the definition in Article 1(1)(b) 2001/83/EC. The sensor head (2) and the display (3) are a device/apparatus for the purpose of diagnosing and monitoring a disease and are therefore the medical device. |
| Integral part | The medicinal product is an integral part of the medical device and the whole product is therefore an integrated product (combination product). |
| Main effect | The main effect is the calculation of the enzyme level using the measuring signal generated by the mixture of substances. |
| Ancillary effect | The change in pressure as a result of the measured value is enhanced by the mixture of substances, making the measurement more precise and more stable |
| Conformity assessment | The MDR is authoritative, as the main effect is in the medical device. The requirements of Annex I of 2001/83/EC continue to apply on the basis of Annex I Paragraph 12.2. |
| Classification | Class III due to Rule 14 |
| Authorities | Notified body (class III authorization) for the medical device |
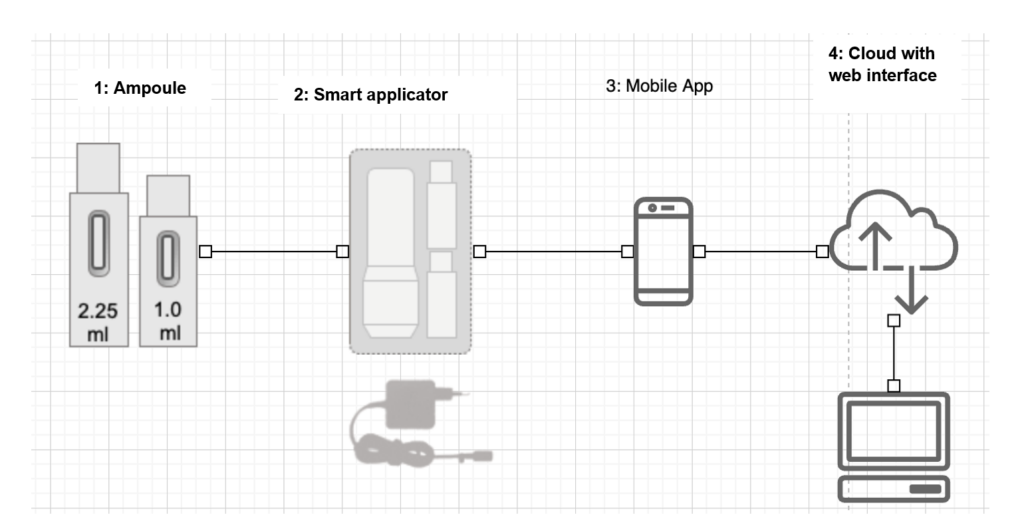
The product is used to provide treatment of a disease with medication. The medication is pre-dosed by manufacturer A and delivered in ampoules (1). The medication is administered using the smart applicator (2), which controls and fully releases the medication and sends the data on the procedure to a mobile app (3). The mobile app (3) records the time series that a doctor can evaluate remotely via the cloud (4). The manufacturer of the smart applicator and the software products is manufacturer B. Manufacturer B packages parts (2) and (1) together. The user can buy additional ampoules (1) in a pharmacy.
Regulatory strategy
| Combined product parts | The ampoule (1) containing the medication is the medicinal product. The ampoule is the primary packaging. The smart applicator (2) is the medical device (not an accessory as accessories are bound to medical devices and not to medicinal products) |
| Integral part | Ampoule (1) is not an integral, permanently installed part of the smart applicator (2), so there is no combination in the sense of the article. |
| Main effect | not relevant |
| Ancillary effect | not relevant |
| Article 1(9) MDR | The smart applicator (2) is intended to release the medication and is therefore subject to the MDR |
| Conformity assessment | Manufacturer A: Ampoule containing medicinal product according to 2001/83/EC Manufacturer B: Components 2, 3, 4 according to the MDR (see Article 1(9)) Manufacturer B must issue a declaration of conformity according to Article 22 for the unit packaged together. The product parts must both be labeled for joint use. |
| Classification | The smart applicator (2) and the software (3, 4) can be classified separately |
| Authorities | Manufacturer A: with EMA/NCA for the ampoule with medicinal product Manufacturer B: with the notified body for the medical device |
6. Summary
The term “combination product” means those medical devices that contain a medicinal product as an integral part. Under EU law, these devices must be regarded either primarily as medical devices or primarily as medicinal products. The rules are derived from Art. 1 (8) and (9) MDR.
a) Medicinal product has priority – 2001/83/EC applies
For combined medicinal products of this type, the MDR integrates its Annex I requirements directly into the Directive for medicinal products by means of Article 117. This change has an impact on manufacturers of combined medicinal products of this type, as a declaration of conformity or EU certificate or opinion from a notified body of the medical device is also needed for the submission.
If the medical device part is affected by a higher classification as a result of the shift to the MDR (the involvement of a notified body becomes necessary), medicinal product manufacturers must request the corresponding certification from the manufacturer of the medical device before the end of the transition periods for class I products.
b) Medical product has priority – MDR applies
Additional requirements in terms of interaction as set out in Annex I of the MDR apply for manufacturers of combined products for which the MDR is authoritative due to the main effect. The requirements laid down in Annex I to Directive 2001/83/EC also apply to medicinal product components. These products are classified in class III in and of themselves. A scientific opinion from the competent authority (EMA) and/or NCAs may be necessary to estimate the risks of the combination.
c) Packaged separately or cross-referenced
Manufacturers that package medicinal products and medical devices together or separately that are intended to be used together must meet the requirements of the MDR together with Article 22 of the MDR.
If you have questions about combination products or need help with the conformity assessment process, please contact the experts at Johner Institute.
We want to thank TÜV SÜD for important input to this article.
Change history
- 2025-11-20: Section 4.d) added (FDA Guidance)
- 2023-09-16: Chapter 4. c) with requirements of FDA for usability added
- 2022-03-01: First version published


