The requirements for clinical investigations to evaluate a device have increased enormously under the MDR. Learn the most important things you need to know about the regulatory pathway for clinical investigations under the MDR here.
Clinical investigation is not the only route to compliance. Alternatively, it can be avoided by taking the right approach to clinical evaluation. The Johner Institute will be happy to advise you on clinical evaluations and check, for example, whether sufficient clinical data is available for your medical device.
1. Clinical investigations of medical devices
a) Definition and objectives
According to the MDR, a clinical investigation is
“any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device”
Source: MDR, Article 2, Paragraph 45
During clinical investigations, medical device manufacturers must prove their devices are safe and deliver the promised clinical performance and benefits – benefits that outweigh the risks. They are obliged to provide this evidence based on clinical data.
Whether a clinical investigation is necessary is usually determined during the clinical evaluation. Here, manufacturers carry out a GAP analysis to uncover any defects in clinical data. If this clinical data is not available in sufficient quantity or quality (e.g., in the scientific literature), manufacturers must collect this data as part of clinical investigations.
Sometimes, a complex and expensive clinical investigation can be avoided by taking the right approach to clinical evaluation. Read more about this in our article on clinical evaluation, or contact the Johner Institute for advice.
b) Differentiation between “clinical investigations” and “other clinical investigations”
The EU Medical Device Regulation MDR has introduced the term “other clinical investigations”.
Clinical investigation
According to Art. 62 Para. 1 MDR, “normal” clinical investigations are those that serve the following purposes:
- a) To verify the suitability of the device for the intended purpose
- b) To review the clinical benefit
- c) To assess the clinical safety and side effects of the device
Manufacturers can use this type of clinical investigation in the conformity assessment procedure. They must fulfill the requirements of Art. 62 to 81 and Annex XV of the MDR.
Clinical investigation after placing on the market (PMCF studies)
In the case of a so-called Post-Market Clinical Follow-up (or PMCF study), however, the determinations from Art. 74 Para. 1 MDR apply. The clinical investigation after placing on the market involves further evaluating devices within the scope of their intended purpose that already bear the CE marking in accordance with Article 20(1).
If the CE marking applies to a different intended purpose, manufacturers must conduct the clinical investigation in accordance with the rules of the “normal” clinical investigation (Art. 62 Para. 1).
You can find out more about Post-Market Clinical Follow-up (PMCF) in our article on Post-Market Surveillance.
Other clinical investigations
According to Art. 82 MDR, other clinical investigations are all clinical investigations conducted for purposes other than those specified in Art. 62 Para. 1 MDR. This also includes basic research and feasibility studies. Such other clinical investigations must meet the requirements of Article 62(2) and (3), (4)(b), (c), (d), (f), (h) and (l) and (6) MDR and, where applicable, other national requirements.
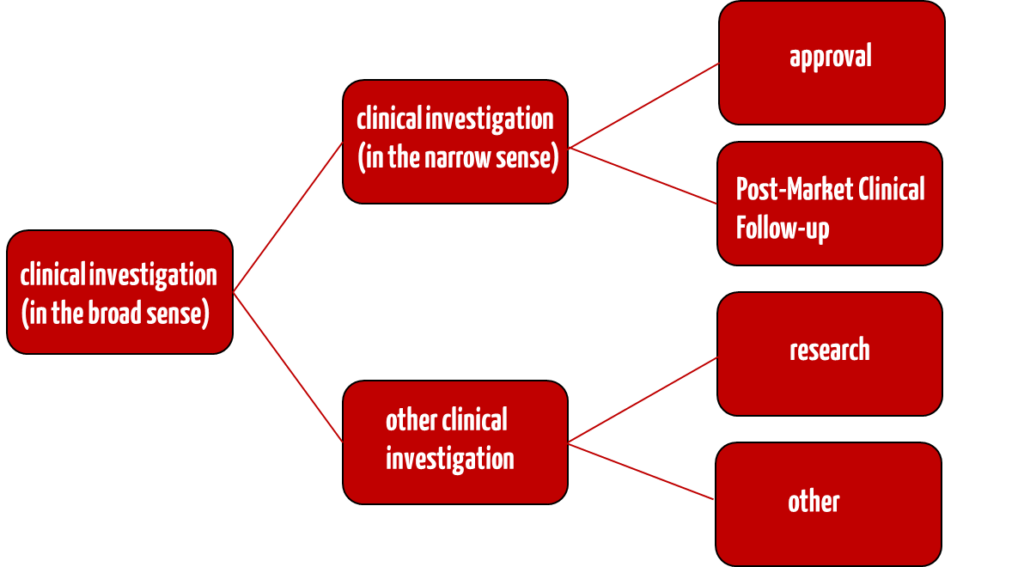
The same regulatory requirements do not apply to clinical investigations and other clinical investigations of medical devices. However, it must be checked whether the BfArM must be involved in both cases. In both cases, the requirements of the MDR must be observed, and, as a rule, an ethics committee must be asked for approval.
The following example illustrates the distinction:
| other clinical investigation | clinical investigation | |
|---|---|---|
| purpose | Other purposes and NOT under the conformity assessment procedure: clinical investigations not conducted for one of the purposes referred to in Article 62(1) MDR (Article 82 MDR). | (a) to determine and review that a device is designed, manufactured, and packaged to be fit for one or more of the listed specific purposes under normal conditions of use and to perform as claimed by its manufacturer; (b) to determine and review the clinical benefits of a device as claimed by its manufacturer; (c) to determine and review the clinical safety of the device and to determine any undesirable side effects of the device under normal conditions of use and to assess whether these represent acceptable risks in relation to the benefits provided by the device (Article 62 MDR). |
| situation | A team of researchers uses an (already approved) MRI device to test, combine, and improve MRI sequences to diagnose a specific cancer more reliably. They are testing these measurement sequences on patients whose cancer diagnosis has already been confirmed or ruled out by histology. | A manufacturer then incorporates the MRI sequences developed by the research team into the next version of its MRI device. Because the clinical data from the research team is not sufficient (e.g., because not enough patients have been examined or the data cannot be transferred 1:1), the manufacturer conducts a clinical investigation with the device and the newly developed sequences to prove the benefit and performance of its new medical device. The manufacturer must do this as part of the conformity assessment (“approval”). |
| evaluation | This “trial” constitutes a “clinical investigation,” i.e., an “other clinical investigation,” and requires the approval of an ethics committee appointed by national law and is therefore subject to approval. | This “pivotal study,” previously also referred to as an “MPG study,” is a clinical investigation as defined by medical device law and is regulated in the MDR in Article 62 (1) et seq. |
Particularly in the case of medical devices based on new procedures, manufacturers find it challenging to make a distinction: Is the purpose of the clinical investigation to try out or develop the procedure? Or does it serve to collect clinical data for the approval of the medical device? Or both? Manufacturers should clearly define this.
2. Categorization of clinical investigations
a) Possibilities of categorization
Clinical investigations can be categorized according to various aspects:
- by purpose (e.g., approval, research, etc., see below)
- by phase (at the beginning of development, before approval, after approval, etc.) The classification into phases 0 to IV is mainly found in drug studies and is not the subject of this study).
- according to study design (see below)
b) Categorization by purpose
The different types of clinical investigations in the field of medical devices serve different purposes. Depending on the purpose, they are subject to different requirements.
| type | objective | date in relation to placing on the market | intervention in diagnosis or therapy |
|---|---|---|---|
| research study (a form of “other clinical investigation”, Article 82 MDR and Section 24 MPDG) | gain insights, determine feasibility | before | yes and no |
| pivotal study (clinical investigation), Article 62(1) MDR) | demonstrate the safety, benefit, and performance capability of an (otherwise finished) device | before | yes |
| PMCF study (clinical investigation with a device bearing the CE marking, Article 74 MDR) | demonstrate the safety, benefit, and performance of a device on the market | afterwards | yes |
| PMCF study without additional stressful and/or invasive procedures (either another clinical investigation or a form of PMCF study) | either for research purposes without proof of safety, benefit, and performance capability or for proof of safety, benefit, and performance capability of a device on the market | afterwards | no |
Examples of “measures”:
- Additional ultrasound examination
- Additional blood test
- Extended physical examination
- Assignment (also randomized) of the patient to a control group that is examined or treated differently from another group
If, on the other hand, medical personnel were only observed at work or patients were interviewed, this would not be considered an intervention or measure. There would be no clinical investigation. In this case, it would be a non-interventional clinical investigation.
c) Categorization according to study design
This categorization is based on the attributes of the study design, for example
- number of patients (< 100, > 1000 (= cohort))
- number of study centers (e.g., multicenter)
- time of planning: retrospective versus prospective
- blinding: unblinded (“open-label”), single-blinded (“single-blinded”), double-blinded (“double-blinded”)
- randomized/non-randomized: Allocation of patients to study arms at random
- with or without the use of placebos (“placebo-controlled”)
- period: Longitudinal studies follow patients over a longer period, sometimes over their entire lives.
- interventional, non-interventional
Different study designs are defined depending on these attributes. The following table gives examples.
| study design | scientific significance | number of patients | number of centers | time of planning | blinding | randomized | period | interventional |
|---|---|---|---|---|---|---|---|---|
| randomized study | very high | several to many | usually several | prospective | yes/no | yes | depending on objective | yes |
| cohort study | high | many | usually several | prospective / retrospective | yes/no | no | years | yes |
| cross-sectional study | medium | many | usually several | prospective | no | no | point in time, possibly several | yes |
| case series | medium to high | several to many | one or several | prospective / retrospective | yes/no | yes/no | depending on objective | yes |
| case study | low | one | one | prospective / retrospective | no | no | mostly point in time | yes/no |
The MDR requires manufacturers to determine the level of evidence. The higher the level of evidence, the higher the scientific significance must be.
The objective of the clinical investigation and the primary endpoint aligned with it determine the design. This results in the evidence and the scientific significance.
A randomized clinical investigation is not always possible. It is also possible to conduct a case series in a prospective and controlled manner and thus achieve level 3. However, this level alone does not determine the scientific significance.
3. Clinical investigations in practice
a) Phases and activities
Clinical investigations of medical devices are usually conducted in phases, ideally sequentially, in practice, and sometimes iteratively.
- Setting objectives
In the first phase, the manufacturers, as sponsors of the clinical investigation, determine the regulatory requirements, decide whether a clinical investigation is necessary, and roughly determine its objectives. - Plan the clinical investigation
The manufacturers then formulate the hypotheses and objectives in more detail – usually with the help of biostatisticians – and determine the study design. They plan the course of the study, select people, and provide budgets. - Prepare the clinical investigation
The manufacturers select the trial centers and clinical investigators and obtain the vote of ethics committees and authorities. They prepare the processes (including documentation) and tools (e.g., electronic data collection) and train those involved. - Conducting the clinical investigation
The investigators and the medical staff involved collect the data. The clinical monitors continuously review the data for plausibility and completeness, as well as the implementation at the trial site during on-site visits. The data managers evaluate the data on an ongoing basis in order to inform the authorities in the event of problems or to be able to adjust or terminate the clinical investigation. - Evaluate data and assess the safety and performance of the device
The manufacturers or their service providers evaluate the data. They assess whether the hypotheses have been verified and, thus, whether the safety and performance of the device have been proven. They prepare reports and the clinical evaluation.
b) Important points for clinical investigations
Annex XV Chapter I of the MDR determines the points to be observed in clinical investigations:
- Recognized ethical principles are observed.
- The inspection is carried out according to a protocol that corresponds to the state of the art in science and technology and is adapted to the device to be tested.
- Testing is carried out under anticipated conditions of use and with the involvement of a representative number of users.
- All characteristics of the device are taken into account and documented.
- The investigator and his staff have access to the technical and clinical data of the device and are adequately instructed in its handling.
- The clinical investigation report critically evaluates all data collected, including any negative outputs.
c) Typical mistakes manufacturers should avoid
The Johner Institute regularly encounters the following sources of error with manufacturers conducting clinical investigations as sponsors:
Lack of clarity about the need for a clinical investigation
The manufacturers do not know whether a clinical investigation is necessary at all. In particular, it is unclear whether the data to date is sufficient to prove safety and performance. An unnecessary inspection is a waste of time and money. If, on the other hand, a clinical investigation is required but has not been carried out, this will hazard legally compliant marketing.
Imprecise or incorrect objectives
Manufacturers must specify precisely which “endpoints” the study must demonstrate with clinical data. Otherwise, they run the risk that the study proves a hypothesis but that this proof is not suitable to demonstrate safety, performance, and clinical benefit.
Incorrect study design
The importance of planning can hardly be overemphasized: The wrong population size, an inappropriate clinical investigation format, or an unrealistic project plan can all contribute to the failure of a clinical investigation. An agile, iterative, incremental approach is also not an effective recipe in the context of clinical investigations.
Inadequate monitoring and data management
Errors and gaps in the data and data collection that do not correspond to the clinical investigation plan destroy the informative value of a clinical investigation. Therefore, close monitoring and sound data management are essential for clinical investigations.
Lack of equivalence
Some manufacturers use the findings from the clinical investigation to improve the device further. They must then demonstrate the equivalence of the devices used in the clinical investigation and the devices that are to be approved.
Insufficient number of participants
Many manufacturers find it challenging to recruit sufficient trial participants to achieve the necessary evidence. This is particularly true in these cases:
- Rare disease
- The advantage (= superiority) of the device is small or even questionable compared to alternatives.
- The device appears strange or “risky” to patients.
- There are not enough investigators interested in the study.
Lack of clarity about regulatory requirements
The MDR and the MPDG have now clearly defined and established the requirements for “other clinical investigations.” This also applies to PMCF studies. Manufacturers should be aware of this. They should also not assume that “studies” with already approved devices do not require approval.
d) Tasks and selection of Clinical Research Organizations (CROs)
Clinical Research Organizations (CROs) can help with the above steps if necessary. However, most CROs specialize in drug research.
Using a (possibly expensive) CRO is not always necessary because not every device needs a clinical investigation or PMCF study. You can learn more about this in our articles on the MDCG 2020-6 and clinical evaluation.
e) Use of digital health tools
The use of digital health tools can significantly increase the speed and quality of, e.g., data collection. However, regulatory constraints must be considered. The following scientific articles provide an overview of these.
- Digital Tools—Regulatory Considerations for Application in Clinical Trials (springer.com)
- Digital Health Technology (DHT) in European Clinical Trials, How to Improve the Status-Quo of the Regulatory Landscape? (springer.com)
4. Regulatory requirements for clinical investigations
a) Medical Device Regulation MDR
The MDR determines its requirements for clinical investigations in Articles 62 to 80 and Annex XV. Overall, the requirements of the MDR are extensive and specific. For example, there are particular requirements for information and inspections for special groups of people, such as children, pregnant women, and breastfeeding mothers.
The MDR also determines when and how data must (in the future) be deposited in EUDAMED, how to proceed in the event of changes to the study design, and what the requirements are for clinical investigations with devices that already bear a CE mark.
Annex XV of the MDR sets out further requirements for clinical investigations’ conduct, documentation, and sponsors (manufacturers).
The MDR also reserves the right to add further requirements via Common Specifications (CS).
The guideline MDCG 2021-6 “Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation” provides an initial remedy. The document is aimed at sponsors of clinical investigations of medical devices that are conducted within the scope of Regulation (EU) 2017/745 (MDR).
b) Medical Device Implementation Act MPDG
The Medical Device Implementation Act (MPDG) has replaced the MPG and the MPKPV. It now imposes almost the same requirements on “research studies” (“other clinical investigations”) and “clinical investigations.” The MPDG also insists on “other clinical investigations”:
- Minimization of risks and burdens and their compatibility with the expected benefits
- Qualification of the investigators
- Insurance cover
- Consent of the test subjects (there are special regulations)
- A positive vote by the ethics committee (exceptions may be possible for CE-marked devices)
- Notification to the competent higher federal authority (exceptions may be possible for CE-marked devices)
For “other clinical investigations,” the MPDG requires that “the other clinical investigation has been notified to the competent higher federal authority in accordance with § 53 (1).” In the case of “clinical investigations,” it is required that “the competent higher federal authority has issued an authorization.”
PMCF studies, in accordance with Article 74 MDR, are not explicitly described and defined in the MPDG. However, manufacturers must comply with the requirements of the MDR.
The BfArM explains in its guidance for sponsors how clinical investigations and clinical performance studies are to be recorded in the DIMDS.
c) MDCG and MEDDEV documents
The EU Commission and the notified bodies have published further documents. These include the following MDCG guidance documents and some MEDDEVs, some of which are still considered to be state of the art and can, therefore be consulted:
- MDCG 2021-28: Substantial modification of clinical investigation under Medical Device Regulation
- MDCG 2021-8: Clinical investigation application/notification documents
- MDCG 2020-10/1 und MDCG 2020-10/2: Guidance on safety reporting in clinical investigations und Appendix: Clinical investigation summary safety report form
- MDCG 2020-6: Guidance on sufficient clinical evidence for legacy devices
- MDCG 2020-5: Guidance on clinical evaluation – Equivalence
- MEDDEV 2.12/2: Post Market Clinical Follow-up Studies
d) ISO 14155
The most precise specifications for conducting clinical investigations are set out in DIN EN ISO 14155: 2021, a standard entitled “Clinical investigation of medical devices in humans – Good clinical practice.” For example, it specifies
- how test plans are to be drawn up,
- how to deal with changes,
- which documents and forms with which content are required, and
- who bears which responsibilities.
e) Further regulatory requirements in the context of medical devices
Manufacturers should observe these regulations and best practices:
- Medical Device User Notification and Information Ordinance (“Medizinprodukte-Anwendermelde- und Informationsverordnung”, MPAMIV)
- Ordinance on the database-supported information system on medical devices of the German Institute for Medical Documentation and Information (“Verordnung über das datenbankgestützte Informationssystem über Medizinprodukte des Deutschen Instituts für Medizinische Dokumentation und Information”, DIMDIV
- IMDRF MDCE WG/N 65 FINAL:2021: Post Market Clinical Follow-up Studies
- FDA Guidances on “Good Clinical Pratice”
f) Regulatory requirements for clinical investigations in the context of research
Suppose the clinical investigation is NOT used to demonstrate safety and performance as part of the approval of a device. In that case, it is classified as “other clinical investigations” (Art. 82 MDR), and therefore the requirements of the MDR apply.
This data cannot be used for the conformity assessment. However, manufacturers can use the data as pre-clinical data for the clinical evaluation.
In addition, the “Good Clinical Practice” rules must also be observed in clinical research, for example. The Declaration of Helsinki must be observed in all cases. In most cases, a vote of the ethics committee must be obtained.
5 Conclusion and recommendation
Understandably, many manufacturers are reluctant to subject their medical devices to clinical investigations. The effort involved and the regulatory requirements are immense. Mistakes can also happen quickly, destroying the value of the clinical investigation or even having criminal consequences.
The MDR increases the requirements for clinical investigations on the one hand and the number of cases in which a clinical investigation is necessary on the other. This is also because the MDR and many notified bodies only accept new clinical data, as the requirements for equivalent devices have increased.
For this reason, the Johner Institute recommends either planning the clinical strategy early on in the development process, especially for innovative devices, implantable devices, and class III devices, or collecting the missing clinical data for CE-marked devices as part of PMCF studies to avoid deviations and, in the worst case, certificate withdrawals.
The Johner Institute advises on documents, content, and the conduct of clinical investigations. We are also happy to advise you on strategies and possible alternatives to clinical investigations.
Change history:
- 2024-11-08: Chapter 3.e) with digital health tools added, numbering of chapter 3 corrected
- 2022-09-23: Guideline of the BfArM for sponsors added
- 2022-09-07: Complete update of the article


