The 510(k), also known as “Premarket Notification”, is one of the most common procedures for the authorization of medical devices in the USA. The concept is based on demonstrating equivalence with a predicate device (equivalent device). But the same mistakes, which can result in the entire 510(k) authorization failing, are made time and again. It doesn’t have to be this way.
This article will explain the five most common mistakes made when submitting a 510(k) and how they can be avoided.
Check out our Medical Device University program.
And find out how a 510(k) process works in our article FDA 510(k): Premarket Submission.

Mistake 1: Identifying the product code incorrectly
Unlike in EU law, where devices are classified on a case-by-case basis by following rules, there are predefined fixed codes for specific device classes. Therefore, manufacturers “only” have to identify the right code for their device from more than 1700 different codes.
The product code provides valuable information for the submission, including:
- The type of submission required (e.g., 510(k), 510(k) Exempt)
- The device class (I, II or III)
- The regulation number with the device description
- Recognized consensus standards for the device
- Requirements for the special controls
But mistakes are often made when trying to identify the right product code.
Solution:
Timing is critical: You should determine the product code when the intended use of your product has been established.
The FDA describes the term “intended use” as follows:
intended use means the general purpose of the device or its function, and encompasses the indications for use.
We use the term “intended use” in this sense in the rest of the document.
The next steps are:
- Step 1: Define the intended use
- Step 2: Search for predicate devices on the market and find out their product codes (in the 510(k) database)
- Step 3: Search in the FDA database itself for product codes using the classification panels
- Step 4: Evaluate the product codes found using the most appropriate characteristics and work out the appropriate product code
The Johner Institute will be happy to help you if you have any questions about the 510(k) procedure. Our experts will, of course, also be happy to answer any questions you may have about product codes. Contact us!
Product code through “Classify Your Medical Device”
The FDA also offers assistance. The website Classify Your Medical Device describes different ways of working out the classification and therefore the product code of your device.
If you have questions: Contact the FDA
If there is anything you are not sure about, you can also contact the FDA.
- without obligation by email to: [email protected] or [email protected] or
- formally via a 513(g) request through which the manufacturer asks for a formal statement or classification of its device by the FDA.
Above all, a 513(g) request prevents a device from being submitted with an incorrect code by mistake. However, the FDA charges a fee for these requests.
The user fee for the 513(g) request is:
- Normal: $5,061
- For small businesses: $2,530
Information on how to submit a 513(g) can be found in the following guidance document: FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act
Mistake 2: Not following device-specific requirements
The FDA has established specific requirements for numerous devices that must be complied with and demonstrated in the 510(k). Not knowing these requirements or failure to fully comply with them often leads to questions from the FDA. For example:
- Device-specific performance standards are not met.
- Insufficient justification is given as to why certain standards do not apply or only apply in part.
- Not enough device tests are performed.
- The special control guidelines are not followed.
- The declaration of conformity is incorrect or does not include all the appropriate recognized standards.
Solution:
First of all, manufacturers should check whether there are device-specific requirements that have to be taken into account in their Premarket Notification. Manufacturers can identify these requirements by:
- classifying their device
- working through the FDA’s special control guidelines
- searching in the FDA guidance documents database for the respective device to identify any guidance documents with information about device-specific requirements
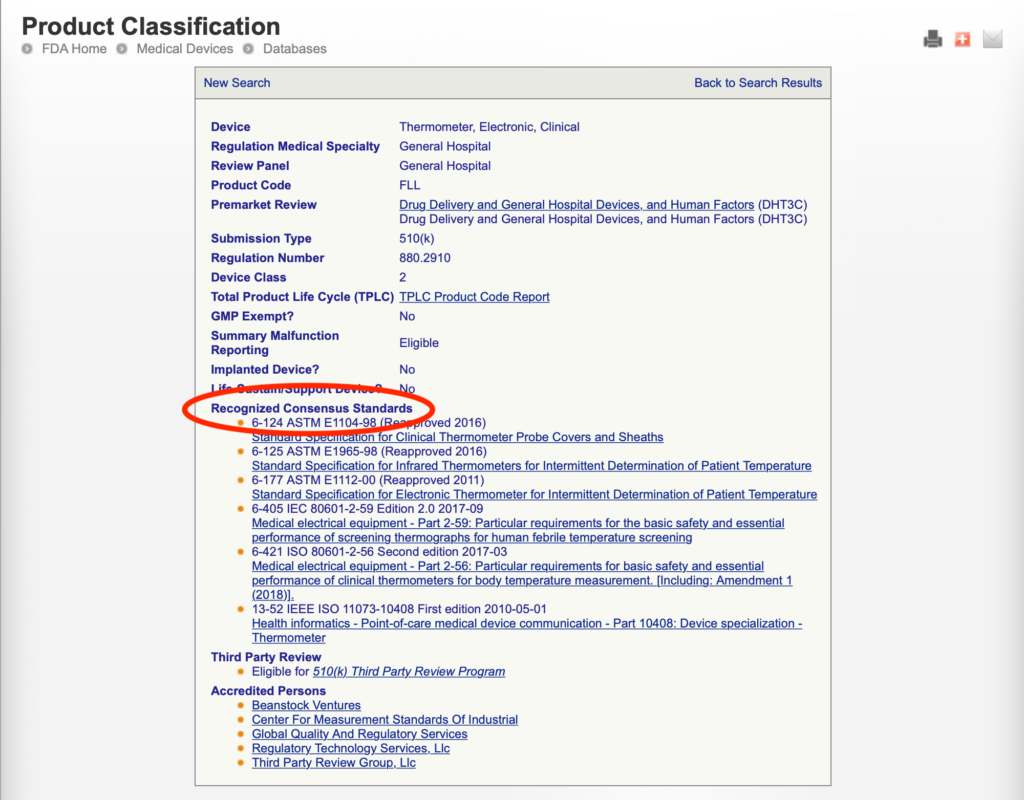
- searching the FDA product classification database to identify any recognized consensus standards that apply to the product code
Example:
Mistake 3: The predicate device is NSE (not substantially equivalent)
Please also have a look at the article on Predicate Devices and “Substantial Equivalence.”
A 510(k) is based on the concept of “Substantial Equivalence” (SE). This means that manufacturers demonstrate that their device is just as safe or safer than an equivalent device already legally available on the market. A device is equivalent (SE) if it has
- the same intended use and
- it has the same technological characteristics (e.g., material, design, energy sources, features).
If the device has the same intended use but different technological characteristics, it must be demonstrated through device tests or clinical or scientific data that
- the device is as safe and effective as the predicate device and
- it does not raise additional questions regarding safety and effectiveness.
The substantial equivalence discussion is therefore an important part of a Premarket Notification. The discussion should include the new device’s characteristics with an explanation from the manufacturer as to how they are comparable to the equivalent device.
Common mistakes include:
- The FDA does not recognize the equivalence of the predicate device because the substantial equivalence has not been sufficiently demonstrated and, e.g., not sufficiently justified. Statements such as “no different questions regarding safety and effectiveness” have to be justified.
- Incorrect use of multiple predicate devices.
While the FDA does permit the use of multiple predicate devices, e.g., if a device combines the characteristics of two predicate devices, all the devices must have the same intended use.
Example: A patient monitor that displays various parameters
The technological characteristics must be comparable. Using the intended use of one device and the technological characteristics of another for the SE comparison is not allowed.
Solution:
These mistakes can be avoided by:
- Selecting a suitable predicate device
The predicate device must be similar to your device with regard to its intended use and its technological characteristics. You can have more than one predicate device, but the FDA recommends defining a primary predicate device.
The following questions will help you identify the right predicate device:
- Does my predicate device have the same intended use?
- Does it have the same technological characteristics (e.g., in terms of design, material, features, energy source, etc.)?
- If the device has different technological characteristics, do they give rise to additional questions regarding the safety and effectiveness of the device?
You can also use the decision tree in Appendix A of FDA guidance document The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] for reference.
- Using FDA guidance as the basis for your substantial equivalence discussion
The FDA has set out its requirements for the substantial equivalence discussion with information on what has to be taken into account with regard to the intended use and technological characteristics in its guidance document The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].
Mistake 4: Formal errors
Formal errors are definitely one of the most common problems with applications of all types. That’s why the FDA reviews the formal requirements in two steps before the 510(k) is even submitted to a technical reviewer:
- Log-in and acknowledgment procedure: To pass this check, the Premarket Notification must comply with eCopy requirements and the user fee must have been paid.
- Acceptance review: In this case, the FDA checks whether the submission is formally complete based on the RTA (refuse to accept) checklist before it is sent to the technical reviewer for the substantive review.
The most common formal errors in 510(k)s include:
- The application does not meet the requirements for the electronic submission format (eCopy format).
- The cover letter does not contain all the information requested.
- The user fee has not (yet) been paid.
- Information is missing as, e.g., the RTA checklist was not followed correctly.
Solution:
- Clarify which 510(k) type you want to use (Special, Abbreviated, Traditional) and submit all the requested information.
- If you aren’t sure exactly which information you need to submit to the FDA, you can submit what is known as a pre-submission request and ask your questions to the FDA directly.
- The electronic submission formatting requirements can be found in the corresponding eCopy guidances.
- You should also make sure you go through the FDA’s RTA checklist before submission.
The tools provided by the FDA are also helpful:
- eSubmitter-eCopies Tool: This tool can be used to create eCopy-compliant submissions.
- eCopy Validation Module: This tool makes it easier to check a submission for compliance with eCopy requirements.
- eSTAR: This is an interactive PDF from the FDA that guides you through the process of preparing a 510(k). The content and structure are predefined. The advantage of eSTAR is that it eliminates the RTA (refuse to accept) check.
Mistake 5: Not following the FDA guidance documents
The FDA itself offers extensive guidance for 510(k)s. Anyone who doesn’t pay (enough) attention to these guidance documents often makes the following mistakes:
- Important documents are missing from the application. Structuring the submission in such a confusing way that the reviewer cannot find information
- Submitting documents that do not contain all the information required by the FDA (incomplete SE discussion, insufficient risk analysis, incomplete software architecture, etc.)
- Providing information in the submission that is inconsistent or contradictory (e.g., different intended uses in different places in the submission)
- Giving an incomplete device description that does not fully correspond to the product code identified
Solution:
The FDA has published guidance documents on what content is required in a Premarket Notification. These include lists of which documents have to be submitted. They should, therefore, be taken into account even in the development stage. This ensures that all the documents required are created.
The following documents are particularly worth paying attention to:
- Format for Traditional and Abbreviated 510(k)s
- For devices that contain software: Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices and the draft document: Content of Premarket Submissions for Device Software Functions
- For usability: Applying Human Factors and Usability Engineering to Medical Devices
Numerous other guidance documents on a wide range of topics are available in the FDA database.
Summary
510(k)s, also known as Premarket Notifications, are the specified pathway to the US market for a lot of devices. However, you must avoid the most common mistakes. In summary, you should pay attention to the following:
- Identifying the right product code
- Classify you device using the panels or the Classify Your Medical Device website
- If you have any doubts, contact the FDA, either by mail to [email protected] or [email protected] or via a 513(g) request
- Defining the device-specific requirements
- After classifying your device, work through the FDA’s special control guidelines
- Search the FDA guidance documents for the corresponding product code. Identify recognized consensus standards with information on device-specific requirements
- The predicate device is NSE (Not Substantially Equivalent)
- Select a suitable predicate device
- Base the substantial equivalence discussion on FDA guidance
- Complying with the formal requirements
- Go through the RTA checklist
- Use FDA tools: eSubmitter-eCopies Tool (create eCopy-compliant submissions), eCopy Validation Module (check compliance with eCopy requirements), eSTAR (interactive PDFs)
- Paying attention to the guidance documents in the FDA database
With the FDA series in our Medical Device University, you can create your 510(k) submission to move through the procedure quickly. Please also refer to our articles on 510(k)s.


