In 21 CFR part 11, the FDA establishes its requirements for electronic records and signatures, which also apply to medical device manufacturers.
A lot of companies print everything out on paper and then sign it by hand to circumvent the requirements of part 11. Is this really necessary?
1. 21 CFR part 11: A source of fear?!
The FDA’s part 11 on “Electronic Records; Electronic Signatures,” published in 1993, has caused many companies sleepless nights, especially in the pharmaceutical sector – and good business for consultants.
Sometimes the requirements were interpreted in such an over-the-top manner that, in 2003, the FDA felt compelled to publish the Guidance document: “Part 11, Electronic Records; Electronic Signatures — Scope and Application” to provide clarity. In the end, the authority saw its own objective, namely to use part 11 to provide a basis for the replacement of paper documents by electronic information, being thwarted.
But what does 21 CFR part 11 really require? And which documents are affected?
2. Affected documents and systems
a) Affected documents/records
21 CFR part 11 applies whenever information is to be electronically generated, amended, stored, transferred, or accessed. This can involve very different types of information, such as:
- Text
- Images, videos
- Audio files
The requirements (for IT systems) must be met if the documents generated, stored, transmitted, or amended are used to demonstrate compliance with regulatory requirements, such as:
- Release and test protocols
- Process and work instructions
- Design drawings, software architecture documentation
- Specifications, request documents
- Records, e.g., production records
- Review protocols
As a rule of thumb, you can say that systems are subject to 21 CFR part 11 if the documents “managed” with the systems are
- submitted to the FDA (e.g., for a 510(k) submission) or
- relevant for an FDA inspection, i.e., the inspection of the QM system to ensure it complies with 21 CFR part 820.
The FDA does not require certain systems to be “part 11 compliant”:
- Old systems that were in operation before August 20, 1997
- Systems that generate paper printouts
So, 21 CFR part 11 is only applicable if electronic records are replacing paper records.
There is a gray area when a system can produce a paper printout but relies on electronic recording to generate it. For example, manufacturers often automatically generate thousands of pages of test reports, print them out, and sign them. In this case, you would have to justify the decision not to apply part 11.
b) Affected systems
Computerized systems that generate these documents must meet the legal requirements, e.g., be documented and validated. Examples of such systems can be:
- Test and test benches
- Software tools for determining code metrics and executing automated tests
- Tools such as Polarion, Jira, and Confluence, which are used to create records, e.g., to complete checklists
- Web-based software that is used to create documents such as specifications
- Production and process systems that log automatically
3. The requirements of 21 CFR part 11
The 21 CFR part 11 comprises three “subparts” (see Fig. 1):
- Subpart A: General provisions, in particular definitions
- Subpart B: Requirements for electronic records
- Subpart C: Requirements for electronic signatures
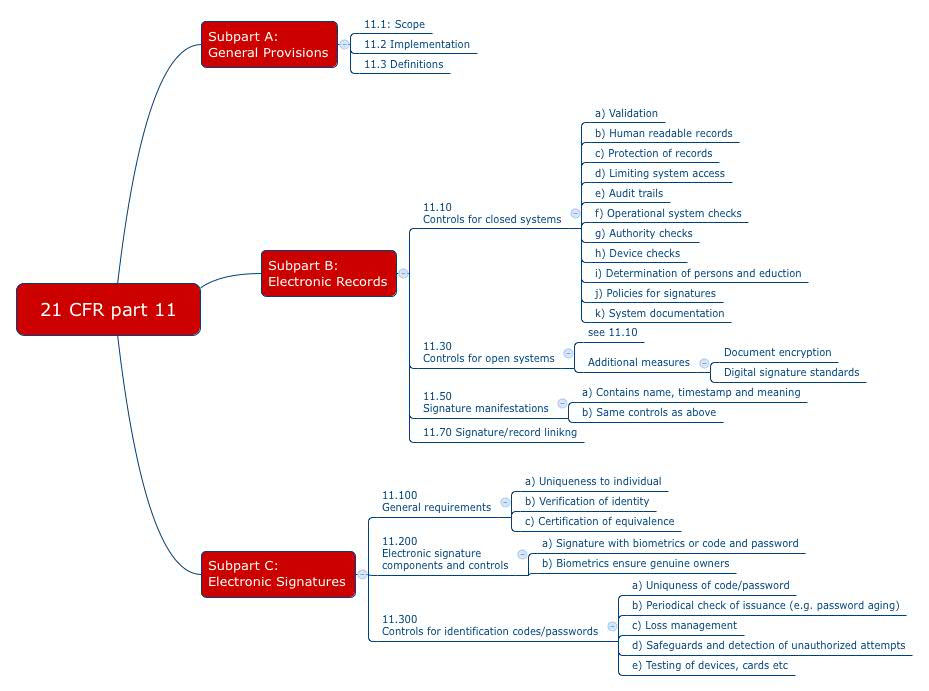
The FDA distinguishes between open and closed systems when it comes to requirements. A system is closed when the system is under the control of persons who are responsible for the electronic records managed by this system. Otherwise, it is an open system.
An example of a closed system would be a build and test system on the intranet that only the testers or developers responsible can access.
A system that transmits data via the internet is also considered an open system.
a) Requirements for closed systems
21 CFR part 11.10 defines the requirements for closed systems. The idea behind the requirements is that the people who work with these systems must ensure the authenticity, integrity and, if necessary, confidentiality of the data. For this reason, the following are obligatory:
- System validation (performance, the ability to detect invalid or altered records)
- Generation (also) of human readable records
- Ensuring the protection of records (must be available)
- Limiting system access to authorized individuals
- Use of computer-generated, time-stamped audit trails that show who changed what and when. But here the FDA is rowing back, as you can read in the above mentioned guidance document.
- Operational system checks to ensure that (only) the permitted sequencing of steps and events is enforced – if necessary
- Authority checks to ensure that only authorized users can use the system (e.g., electronically generate and sign documents), and access the operating system, computer, or peripherals
- Peripherals check to ensure that the inputs and outputs are correct
- Training of the people who work with the system or develop it
- Prevention of falsification so that people are liable in writing for what they sign
- System documentation, e.g., on who has access to the system, how this access is granted, whether it be for the use or maintenance of the system, and on who changed what in the system and when
The FDA requires the IT systems discussed above to be validated and in this context also refers to the “General Principals of Software Validation” guidance document. This leads to the discussion as to whether this is just about validation or about the complete software life cycle. It is the latter that is meant.
Read more about this topic here:
- What is Computerized Systems Validation (CSV)?
- What is software validation?
b) Requirements for open systems
21 CFR part 11.30 places additional requirements on open systems. These include measures such as document encryption and the use of digital signature standards to ensure the authenticity, integrity, and confidentiality of records.
c) Digital signature requirements
The requirements of 21 CFR part 11 regarding digital signatures will seem familiar to anyone who has dealt with this issue before and, for example, the German Signature Act:
- Content: A digital signature must contain
- the name of the signatory,
- the date and time of the signature, and
- the meaning of the signature (e.g., review, approval, author).
- Protection against falsification: It must not be possible to falsify the digital signature (21 CFR establishes the same de facto requirements as are in place for documents).
- Link to document: The signature must be linked to the document in such a way that it cannot be used on other documents.
- Uniqueness: Naturally, it must be possible to assign the signature to a specific individual.
- Biometric and non-biometric methods: The identification must be based on biometric methods or two distinct identification components such as an identification code and password.
When using identification codes (e.g., user name, initials, or number) and passwords, 21 CFR part 11 establishes the following requirements in 11.200 (a) and 11.300:
- Four-eyes principle: The electronic signature must be regulated in such a way that any attempted misuse of someone else’s electronic signature requires the collaboration of two or more individuals.
- Unique combinations: The duplicate assignment of codes and passwords must not be possible.
- Updating: Both codes and passwords must be regularly checked to ensure that they are still sufficiently secure.
- Loss management: In the event that codes, passwords, cards, etc. are lost, there must be a procedure that permits “deauthorization.”
- Security measures: Suitable measures must be in place to protect against and detect unauthorized access attempts.
- Testing: Input/output devices, including cards that bear or read authorization information, must be tested periodically to ensure that they are working correctly.
4. Frequently asked questions regarding 21 CFR part 11
a) Are there any solutions that guarantee compliance with 21 CFR part 11?
The simple answer is no. This is because 21 CFR part 11 doesn’t just establish technical requirements; it also established organizational measures. And you can’t buy those.
However, manufacturers such as our sister company Medsoto have prepared the products in such a way that the technical requirements for creating (technical) documentation are met.
b) Do I have to comply with 21 CFR part 11 if I print everything out and then sign it?
The answer (in most cases) is no. However, there are exceptions, such as the example of test documentation we described above.
c) Do I even have to stop using paper?
The FDA (increasingly) requires you to submit your documents electronically. One example is the eStar format. However, you could also scan and submit printouts. This would allow you to ignore part 11, except for the above exception.
d) What do I do if I have no documents but structured data?
More and more manufacturers, authorities, and notified bodies understand the advantages that structured data has over documents, such as
- freedom from redundancy and, therefore, no inconsistencies,
- automatic verifiability through algorithms, and
- the flexibility to export the data in various target formats for international authorities and notified bodies.
The same requirements apply to this structured data, for example:
- Validation of the systems
- Readability of the data
- Authorization concepts and specifications for approvals
- Protection of the data
- Signatures
These requirements can be met without compiling classic documents such as Word Files or PDFs. Thus, 21 CFR part 11 does not prevent working with structured data.
(Structured) data that manufacturers, authorities, and notified bodies exchange is “serialized,” for example, in JSON or XML format. These data streams correspond to documents that must meet the requirements of 21 CFR part 11, such as being readable or allowing changes to be recognized.
e) What is the best way to meet the requirements for electronic signatures?
Scanned signature: That’s not enough
A first thought might be to scan a signature, insert it into the document, and print it as a PDF. However, this would not meet the requirements of part 11.70. After all, you could export this graphic as a screenshot and insert it into another document.
Using suitable systems: Usually, the ideal solution
Instead, a check digit of the document (hash code) is usually encrypted with the signatory’s private key. This encrypted hash value is the digital signature.
Computerized systems, such as document management systems, must be able to generate this hash code (this is not perceptible to the user).
In 21 CFR part 11, the FDA does not require a fully qualified signature including a certified trust service center. An advance signature level should be sufficient in most cases.
Signature service providers: Compliant but with disadvantages
Other manufacturers use service providers to sign documents. However, this also has disadvantages:
- These services cost money (for every signature).
- This regularly slows down the workflow.
- The service providers rely on documents. This means their concept does not harmonize well with structured data.
The article on electronic and digital signatures provides further tips and explains the required signature level.
5. Conclusion
The requirements of 21 CFR part 11 are comparable to the requirements that medical device manufacturers must also fulfill in Europe. However, some additional requirements such as those relating to audit trails must be fulfilled. The FDA has, though, rowed back a little in precisely these areas with its guidance document.
An advanced electronic signature fulfills the FDA’s requirements for electronic signatures. Manufacturers of medical devices and software can achieve this level of signature without external “trust service providers.”
Common sense should also be applied to electronic records and signatures: Data must not be changed without being noticed, especially if it has already been approved. You want to know who approved this data and when to evaluate the conformity of processes.
That’s all the FDA is concerned with in 21 CFR part 11.
Change history:
- 2024-08-06: Article largely revised and structured, mind map replaced
- 2015-11-13: First version of the article


