The risk management plan is one of the most important documents in technical documentation. Accordingly, authorities and notified bodies examine this plan intensively.
However, it is not only from a regulatory perspective that medical device manufacturers benefit from a precise risk management plan. This article
- lists the regulatory requirements and
- shows how manufacturers benefit from a precise plan.
1. What a risk management plan is
In a risk management plan, a manufacturer documents information on procedures, activities, methods, and tools for the risk management of a specific device throughout its entire life cycle.
This information is usually available as a controlled document. It is also possible to store this information as structured data, as with the real-time compliance system.
Chapter 3 of this article lists the contents of a risk management plan.
2. What advantages the risk management plan offers
Advantage 1: Regulatory compliance and fast approval
Standards such as ISO 14971, Chapter 4.4, and laws require risk management plans. For example, the MDR and IVDR have identical wording in Annex I Section 3:
In carrying out risk management manufacturers shall: (a) establish and document a risk management plan for each device
MDR Annex I, Section 3
If manufacturers meet this requirement, they avoid regulatory hassles with approvals, inspections of technical documentation, and audits, for example, and possible consequences such as delayed approval.
Advantage 2: More efficient development
If a plan is in place, everyone involved knows what needs to be done and when. This is particularly important for successful risk management, to which many different roles contribute:
- development
- risk manager
- quality management
- clinical expert team
- top management
- customers and users
A plan ensures that all activities are coordinated and take place at the right time. It helps to avoid uncoordinated activities and blind performance.
Advantage 3: Better devices
If all risk management activities are carried out by the right people, using the proper methods and at the right time (this is precisely what the risk management plan determines), these activities are particularly effective. This means that risks are identified and controlled with particular reliability. This results in safe devices.
The risk management plan is also necessary in order to derive the product-specific requirements for risk management from the often generally applicable standard operating procedure for risk management. Only the product-specific activities lead to the safety of the device being optimized.
The general risk management plan requires risks to be identified and alternative technologies and architectures to be evaluated during the development of the device.
The risk management plan for standalone software requires that the software libraries used during development must be selected depending on the speed with which their manufacturer delivers patches, among other things.
Advantage 4: Better processes
The risk management plan also helps at the meta-level: it is part of the PDCA cycle (“Plan-Do-Check-Act”). Without a plan, nothing can be done (“Do”) and checked (“Check”). The deviation from the status quo to the plan results in the necessary actions (“Act”) to improve not only the specific plan but also the entire risk management process.
The risk management plan requires that security vulnerabilities in software must be avoided during its architecture phase and searched for during the system test phase by means of penetration tests.
It turns out that the penetration tests identify many vulnerabilities that could have been avoided during the architecture phase.
For this reason, the manufacturer of software products adds the requirement for threat modeling during the architecture phase to its standard operating procedure for risk management.
3. What the risk management plan contains
The minimum requirements for the content of risk management plans are set out in
- the ISO 14971, Chapter 4.4 and
- the position paper of the Team-NB from 2023 (see p. 18).
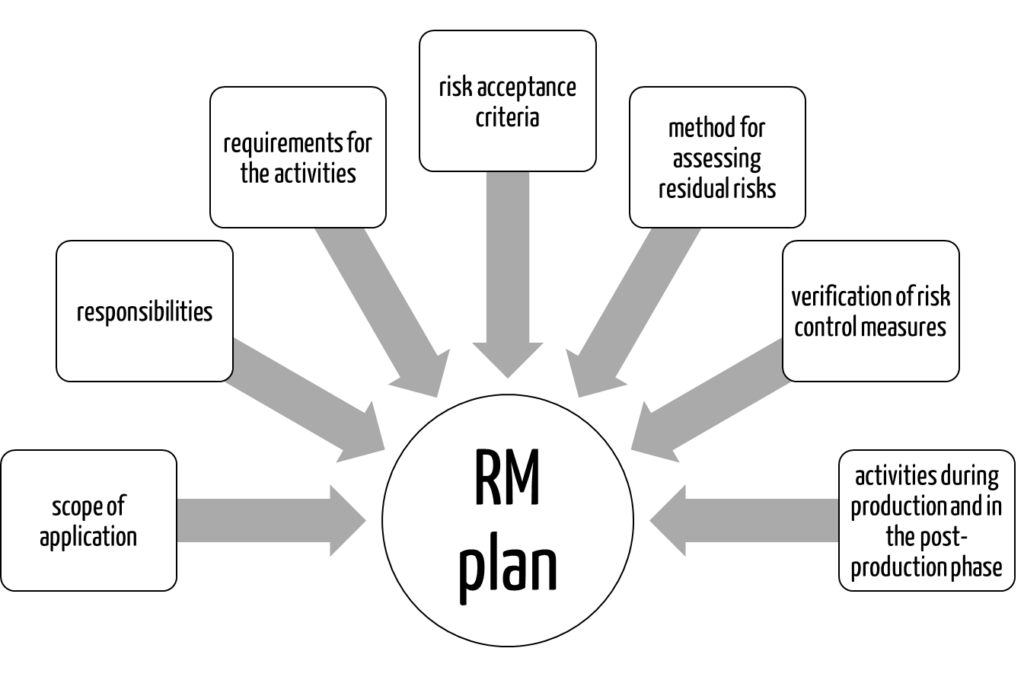
ISO 14971 requires the following elements:
- scope of risk management activities
- responsibilities
- requirements for the activities
- criteria for risk acceptance
- method for assessing residual risks
- activities for verification of risk control measures
- activities during production and in the post-production phase
The following sections contain further information.
a) Scope of application
The scope of application has various dimensions that the risk management plan should consider:
Organizational structure
The persons and departments affected by the document must be specified here. These should also be involved in the creation and release of the risk management plan.
Suppliers carry out many activities in the development and production of medical devices. The plan must, therefore, regulate whether it also relates to them.
Object
The plan should specify which device or devices it relates to. In the case of systems, this can also relate to only part of the system, possibly only accessories for a device or consumables.
Make sure that the interfaces to other devices or components are also aligned with the interfaces of the documents (in this case, plans).
Workflow organization and processes
It should be clarified which processes and life cycle phases the plan covers. Does it only apply to the initial development or also to later design changes? Does it cover the post-market phase?
There is usually a separate post-market surveillance plan, which is referenced in the initial risk management plan.
It should be noted that the post-market plan, according to MDR, does not include the manufacturing phase, whereas the risk management plan, according to 14971, does.
b) Responsibilities
Risk management is a team sport. Typical roles have already been mentioned above. The risk management plan ensures that no roles are forgotten and that all necessary competencies are available.
- In Chapter 7.3.2 on development planning, ISO 14971 requires the manufacturer to define “the necessary resources, including the necessary competence of personnel.” These specifications are specific to the respective development project. Accordingly, manufacturers must define these competencies and provide evidence.
- The “assignment of responsibilities and authorities” is regularly misunderstood. An “authority” has the right to prevent product release during the risk management review if there are doubts about the risk or the benefit-risk ratio. This authority must be determined.
c) Requirements for the activities
Methods and procedures
Various activities must be carried out as part of risk management, e.g., hazard analysis, risk evaluation, definition of risk-minimizing measures, and review of their effectiveness.
Manufacturers must define requirements for each of these activities. This is usually done by defining methods, for example, the PHA for the hazard analysis.
If a higher-level process or standard operating procedure already specifies this, the risk management plan can overwrite it or specify it in another project-specific way.
- The risk management plan specifies that a modular D-FMEA may only analyze the effects of failures up to the “device edge,” i.e., up to the violation of the product specification, using a different assessment, e.g., in the form of an RPN.
- The risk management plan defines the procedure to be followed if the probability cannot be estimated. It determines that, in this case, a worst-case assessment must be made, or the acceptability of the risks must be determined only based on the severity of the risks.
Further specifications
The risk management plan can provide answers to the following questions:
- How is the analysis broken down into modular sub-analyses (design, manufacturing, software, cybersecurity, suppliers, AI/ML, components, lifecycle phases, etc.)?
- In what form are the activities documented?
- Who benefits from which output documents and when?
- Which tools are to be used for the activities?
d) Risk acceptance
Further specifications of the risk management plan concern the risk acceptance criteria, for example,
- the definition of the probability and severity axis,
- the derivation (or acceptance) of the quantitative or qualitative benefits,
- the procedure for assessing the residual risk, e.g., based on the individual risks or at the level of the overall risk.
Please note our tips on deriving risk acceptance.
e) Further specifications
The risk management plan regularly contains references to documents that also apply and must be observed.
4. Typical audit deviations
The Johner Institute’s risk management team has compiled frequently occurring complaints from authorities and notified bodies regarding the risk management plan:
- The team is not complete. For example, an expert for ISO 14971 or a doctor is missing.
- Some auditors and reviewers claim that an acceptance matrix should no longer have three colors. Others state that colors are no longer permitted at all. Neither is true. But generally acceptable risks, which are often described as green, are not permitted by ISO 14971, MDR, or IVDR.
- The risk management plan is completely missing. A standard operating procedure (SOP) alone is not sufficient if it lacks product- and project-specific specifications.
- It is not described how the review is carried out or who participates in it.
- There is no statement on the final overall residual risk or benefit-risk assessment and, in particular, how and with which method this is carried out.
- It is not clear which system or product components the plan covers. Or the plan does not cover the variants of the device and its accessories, consumables, and options.
- Certain aspects are missing from the risk analysis (e.g., manufacturing, software, packaging, storage/transport, accompanying documents, biological safety, etc.) because they were already missing from the plan.
5. Tips for creating the risk management plan
First, manufacturers should ensure that they avoid the mistakes mentioned in Chapter 4.
a) Rule out typical errors
Make sure that you can rule out the deviations mentioned above in Chapter 4 that are identified during audits.
b) Controlling of the risk management plan
A risk management plan is not a static document. It is revised and must be updated continuously as new information becomes available. These revisions should be attached to the risk management file.
c) Review of the risk management plan
A manufacturer should check whether the risk management plan specifies all the risk management activities described in ISO 14971:2019 in Chapters 5 to 10. For each of these activities, the risk management plan should specify:
- the persons responsible for implementation and inspection
- documents in which the activities are documented
- the phase of the life cycle or specifically of the development process in which this activity is carried out
- the aids and tools that will be used in the process
A representative of top management should approve the RM plan to ensure consistency with the established risk policy.
d) Use of the plan
Design reviews can be used to check whether risk management is being carried out in accordance with the plan. The plan also provides the input for the final risk management assessment.
In both cases, it must be checked whether
- the intended persons carried out
- the planned activities
- at the planned time
- with the planned methods and tools and
- have documented them as intended.
This means that the structured and systematic approach to the analysis should not only be planned but also documented accordingly. This has a positive effect on the efficiency of the implementation, prevents gaps and deviations, and also serves later as proof that a systematic approach was actually taken.
A manufacturer shows the auditor the risk table as part of the technical documentation. The auditor looks for three specific risks in the table but does not find them. He hypothesizes that a lack of structure and system is the reason for the absence; therefore, he suspects even greater gaps and ultimately refuses the certificate.
Because the manufacturer has not documented its procedure anywhere, it cannot prove the opposite and has to go through the analysis again in full.
6. Summary and conclusion
A manufacturer must draw up a risk management plan for each device. Standards and laws define the contents of these plans.
Like all plans, the risk management plan must also specify who does what, when and how, and which inputs are converted into which outputs. Like all plans, the risk management plan is not a static document but can be developed further. This is because risk management does not end with development.
It is important to fulfill the minimum requirements of the plan and stick to it later. In this way, manufacturers can not only avoid problems in audits and approvals. A risk management plan is an important building block for the efficient development and production of safe medical devices.
The Johner Institute offers all the support you need to develop and implement audit-proof risk management plans:
- The Medical Device University provides proven templates for a risk management plan.
- In the risk management seminar, participants receive tips and learn tricks for creating plans and checking them for compliance.
- Our experts will review your plans from an auditor’s perspective. This will help you avoid surprises during audits and approvals.
- The risk management team supports you in creating your product-specific risk management plan.
Contact us for a free consultation, during which the experts will give you some initial practical tips.