In this article, you will learn about the requirements that the FDA places on human factors engineering (HFE), the products to which the FDA applies these requirements, and how you can implement these requirements.
1. Human Factors Engineering: When You Need to Comply with FDA Requirements
The FDA’s statements on whether usability engineering should be performed for every medical device are somewhat contradictory.
a) HFE required if product is not Class I or if it contains software
The FDA derives the requirements for human factors engineering from the Quality System Regulations (QSR) described in 21 CFR part 820:
- Design Input: “Address the intended use of the device, including the needs of the user and patient.”
- Design Validation: “Ensure that devices conform to defined user needs and intended uses. This shall include testing of production units under actual or simulated use conditions. Design validation shall include software validation and risk analysis, where appropriate.”
Although the QSRs do not mention human factors engineering by name, the FDA takes this view (see here). The Quality System Regulations apply to every medical device. However, for Class I products, the design controls and thus the two points mentioned above can be largely dispensed with.
Again, there is an important exception: For software, the design controls must be complied with, as the FDA writes in 21 CFR part 820.30 (2) (i).
b) HFE necessary “where appropriate”?
In the preamble to the QSRs, the FDA writes in section i.72. …”when designing a device, the manufacturer should conduct appropriate human factors studies, analyses, and tests from the early stages of the design process until that point in development at which the interfaces with the medical professional and the patient are fixed.”
c) HFE necessary if serious harm can be a consequence of a use error
In its guidance document on human factors engineering, the FDA writes „CDRH believes that for those devices where an analysis of risk indicates that users performing tasks incorrectly or failing to perform tasks could result in serious harm, manufacturers should submit human factors data in premarket submissions (i.e., PMA, 510(k)).“
d) HFE is required if explicitly requested for product category
The FDA explicitly requires human factors engineering in a separate guidance document for several product categories, including ventilators, dialysis machines, infusion pumps, and defibrillators.
e) HFE required if requested by the FDA
The FDA also reserves the right, regardless of the above, to request human factors engineering data for approvals, for example
- if devices have been modified
- if there have been problems
- with specific approval procedures (PMA?)
- if the user group changes.
Conclusion
HFE activities may be less extensive for Class I medical devices. However, risk analysis must be used to justify any decision to omit parts of human factors engineering for all products. Omission is not permitted except in justified cases for the product categories listed under d).
Quite complex? The FDA probably agrees. That is why it recommends contacting them through a presubmission before the actual submission and, if necessary, before the device has been fully developed.
2. Regulations relating to human factors engineering
The FDA refers to several documents relating to human factors engineering:
- The previously mentioned design controls of 21 CFR part 820.30
- AAMI/ANSI HE75:2009 entitled “Human Factors Engineering – Design of medical devices”
- IEC 62366:2015-1, which regulates the usability process, documentation, and training
- ISO 14971:2007 (sic!)
- Guidance Document “Applying Human Factors and Usability Engineering to Medical Devices”
Read more about the US’s regulatory requirements for human factors engineering here.
3. Selected requirements of the FDA
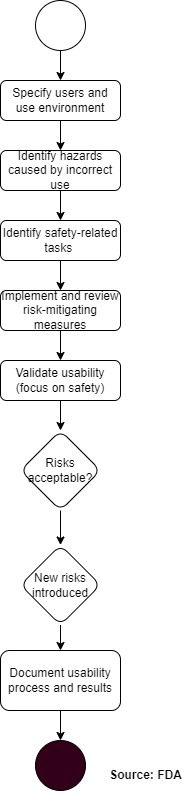
The FDA proposes an approach that is very similar to a standard risk management process:
a) Analyze risks
The first step is to identify safety-related tasks on the system. This focus on tasks (and not, for example, on individual UI elements) is in line with the thinking behind IEC 62366-1:2015.
The FDA lists the following methods:
- FMEA, FTA
- Task analysis
- Expert review
- Context analysis
- Interviews
- Formative assessments, e.g., in the form of a cognitive walkthrough
The FDA also wants the following sources to be taken into account in the risk analysis:
- Feedback from customers
- Feedback from service, sales staff, and trainers
- Results of previous HFE studies
- Technical articles
- Reports to the authorities (including the FDA, of course).
b) Minimize risks
As usual, the risks must be mitigated by the following measures:
- Inherently safe design
- Protective measures
- Instructions
The FDA does not go into too much detail, but as expected, it requires that the effectiveness of the measures be verified.
- The FDA does not consider risk minimization through instructions in the instructions for use acceptable unless the effectiveness can be proven. The same applies to the measure “additional training.”.
c) Validation
The FDA is more specific when it comes to validating usability. It requires that
- All critical tasks are subject to validation.
- Validation is based on previously defined success criteria and demonstrates that no serious usability problems occur.
- Validation includes objective data (user observation) and subjective data (user surveys).
- The final product, including labeling (e.g., instructions for use, labels), is validated.
- Validation is done with representative users (not manufacturer employees) in a representative use environment. Representative also means that the training of the test users must correspond to what a representative user receives.
- At least 15 representatives per user group are involved in the validation.
- The users come from the USA.
We validate the usability engineering of your medical device with US users so that you comply with FDA requirements and are sure to obtain approval.
d) Documentation of the Human Factors Engineering Report
The FDA recommends the following structure for the report:
- Summary
- Description of users, use environment, and training
- Description of the user interface
- Summary of known usage problems
- Analysis of “usage risks”
- Summary of preliminary analysis and evaluation
- Description of critical tasks
- Details of validation
Change history
- 2025-05-20: Editorial changes
