What is the difference between verification and validation, and how are these terms defined? Even standards and regulations use the terms incorrectly or misleadingly.
This article
- provides definitions and examples of the terms verification and validation and
- explains how precise verification and validation of medical devices help with approval and audits.
1. Verification
a) Definition
“Confirmation, through the provision of objective evidence, that specified requirements have been fulfilled.”
ISO 9000
This definition does not explain what type of “requirements” need to be confirmed by verification. Limiting these requirements to product or component requirements is recommended to avoid any uncertainties and confusion with stakeholder requirements.
“Verification is a test using objective evidence that specified characteristics (e.g., of products, components) have been fulfilled.”
ISO 9000
These characteristics or features may be specified in a System Requirements Specification (SRS) or the product requirements specification, respectively.
b) Examples
Examples of characteristics to be specified are:
- Specification of the user interface, e.g., with mockups or drawings (see next chapter)
- Behavior of the system at the data interfaces (including interoperability)
- Behavior of the system at “patient interfaces” (applied part): For example, it could be specified that a certain voltage must be applied to a defibrillator in a certain pulse sequence. The verification would then be the test of whether this voltage is actually present in the specified pulse sequence.
c) Special case: Verification in the context of usability
Verifying the usability represents objective evidence that specified ergonomic features are met concerning usability. Thus, it is a subset of the overall verification process and is required by IEC 62366.
Examples for specified ergonomic features are font sizes, colors, contrast ratios, or general rules such as marking compulsory fields. The verification is carried out, e.g., during formative usability tests, e.g., usability experts check against
- specified product features (e.g., as described in a style guide or a UI mockup) and/or
- general rules such as those described in extensive detail by the ISO 9241 family.
2. Validation
a) Definition
“Confirmation, through the provision of objective evidence, that the requirements for a specific intended purpose or application have been fulfilled.”
ISO 9000
Whether or not these requirements have been met for a specific intended purpose depends on the users and the use context. For example, it is possible that the requirements of a defibrillator are met in an OR setting by professional users but not when laypersons use the device in an emergency on a wet street at night.
The definition of the term “validation” should, therefore, be expanded:
“Verification that the specified requirements are adequate for an intended purpose and the specified users in the specified use context.”
Johner Institute based on IEC 62366-1
b) Examples
Thus, validation is the objective evidence that a specified user can achieve the product’s intended purpose in the specified use context. A validation study has two aspects:
- Classic validation: Testing whether the intended purpose can be achieved with the medical device.
Clinical evaluations are an example for such a validation. A clinical performance evaluation is carried out for in vitro diagnostic medical devices. The clinical evaluation or performance evaluation is part of the validation and must demonstrate that the intended use objectives are achieved, i.e., that the device is safe, efficient, and effective. - Validation of the usability: This is the test of whether the specified users can achieve the intended purpose effectively, efficiently, and satisfactorily in the specified context of use. This test usually takes the form of a summative evaluation (usability test).
3. Software verification and validation
a) Regulatory requirements
The MDR and the IVDR both require:
“For devices that incorporate software or for software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation.”
MDR Annex I Paragraph 17.2.
b) Fulfilment of the regulatory requirements
Manufacturers generally apply IEC 62304 to prove that these verification and validation requirements are met according to the state of the art. However, IEC 62304 only explicitly addresses the verification and refers to IEC 82304-1 for validation.
Verification
Software verification is usually carried out by means of:
- Unit tests
- Integration tests
- Software system tests (see also overview article) in which black box testing methods are used
- Code reviews
- Static code analysis
Validation
The most important methods for the validation of (standalone) software are:
- Validation of usability (summative usability evaluation), typically through usability tests
- Clinical evaluation, which may require clinical investigations
The guidance document on MDCG 2020-1, which we summarized in our article on clinical evaluation of software, provides a valuable addition.
The fastest and most cost-effective way to bring your medical device to market
Our video trainings and numerous templates provide comprehensive guidance on collecting, specifying, and evaluating the requirements for your medical device. The step-by-step instructions make it easy to keep track and go through all the necessary steps.
The Johner Institute can also assist with your software documentation and regulatory compliant development.
4. Common pitfalls
a) Mixing up the various “validations”
The old Medical Device Directive (93/42/EEC) required:
“For devices which incorporate software or which are medical software in themselves, the software must be validated according to the state of the art taking into account the principles of development lifecycle, risk management, validation and verification.”
MDD supplemented to include 2007/47/EC
This requirement uses the term “validation” and “validated” in two different contexts, which should not be confused.
Software validation in the narrow sense
This means the validation described above and should be understood as a delimitation from verification. It tests whether the intended purpose is achieved, and the stakeholder requirements are met.
Software validation in the broad sense
This validation corresponds to Computerized Systems Validation (CSV), or that which the FDA sets out in the guidance document “Software Validation”. Here, the term software validation is used as a synonym for software quality assurance. The latter comprises the life cycle phases:
- Definition of the requirements
- Specification of the software architecture and the software design
- Implementation of the software
- Software testing (unit, integration, and system tests)
- In the case of standalone software, also validation in the narrow sense
An example of software validation in the broader sense is Computerized Systems Validation (CSV).
Our article “Validation of artificial intelligence containing products across the regulated healthcare industries” provides a detailed presentation of verification and validation. We introduce the concept of validation in different contexts: AI, software validation, medical device software validation, and validation in the pharmaceutical context.
Read more about Computerized Systems Validation (CSV) here.
b) Waiver of verification in safety class A
According to IEC 62304:2016, the software safety class determines the need for software system tests necessary to verify the software. No tests are set out in IEC 62304:2006 for safety class A; no unit, no integration, and no system tests. However, such software development would not comply with the MDR requirements.
The IEC 62304 requirements changed with amendment I: IEC 62304 now requires at least software system tests.
According to ISO 14971, all measures contributing to risk control must be verified. If a manufacturer implements measures in software, they are subject to verification regardless of the safety class.
c) Mix-up of verification and validation
In software development, the V-model is still anchored in many people’s heads and referenced by IEC 62304 and IEC 60601-1. But how does IEC 60601-1 understand the V-model?
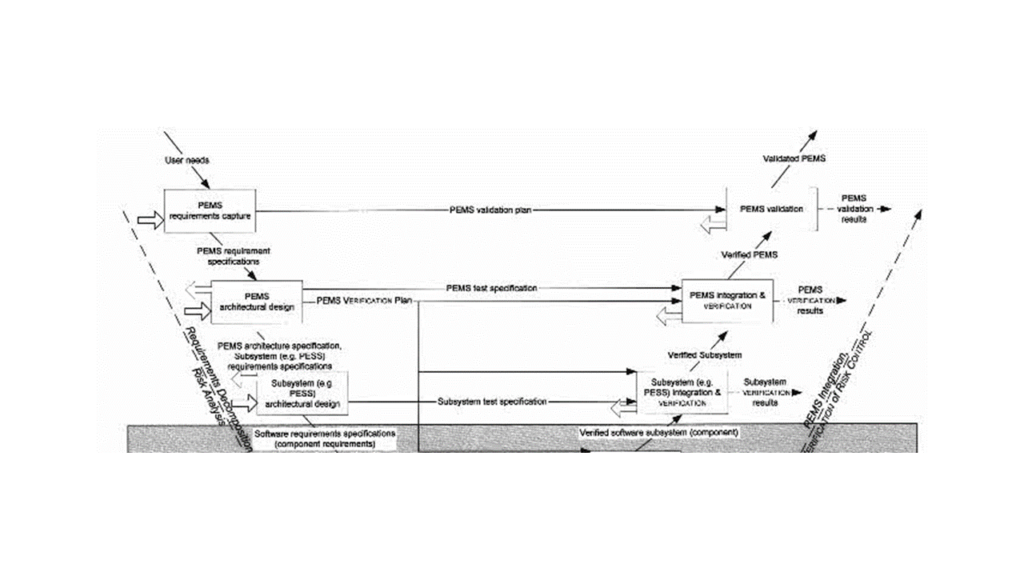
According to the standard, the requirements of a Programmable Electrical Medical System (PEMS) are derived from user needs; in other words, system requirements (see Fig. 1).
The standard does not define “user needs.” The authors probably understand this as an unspecified mixture of requirements, wishes, user requirements, and directly formulated system requirements.
If you are so imprecise with the terms, you will not achieve precise outputs later on.
You can see where this lack of precision leads: The standard believes that fulfilling PEMS requirements, i.e., system requirements, can be validated.
The fulfillment of system requirements can be verified but not validated.
Per definition, you can validate use requirements, but they are not mentioned and get lost in the shuffle of user needs.
d) Misbelief that successful verification is a requirement for successful validation
The results of verification and validation can be independent of one another. It is entirely conceivable for the verification of the medical device to be successful and the validation not or vice versa, as the following examples show:
- The specified 3000 V is generated on the pads of a defibrillator (verification successful). The patient’s heart is not beating (purpose) after using the device (validation failed).
- Instead of the specified 3000 V, only 200 V is generated on the pads of the defibrillator (verification failed). The patient’s heart is beating (purpose) after using the device (validation successful).
e) Verification beyond the device or its components
Manufacturers do not only have to verify the medical device and its components (see right-hand side of the V-model).
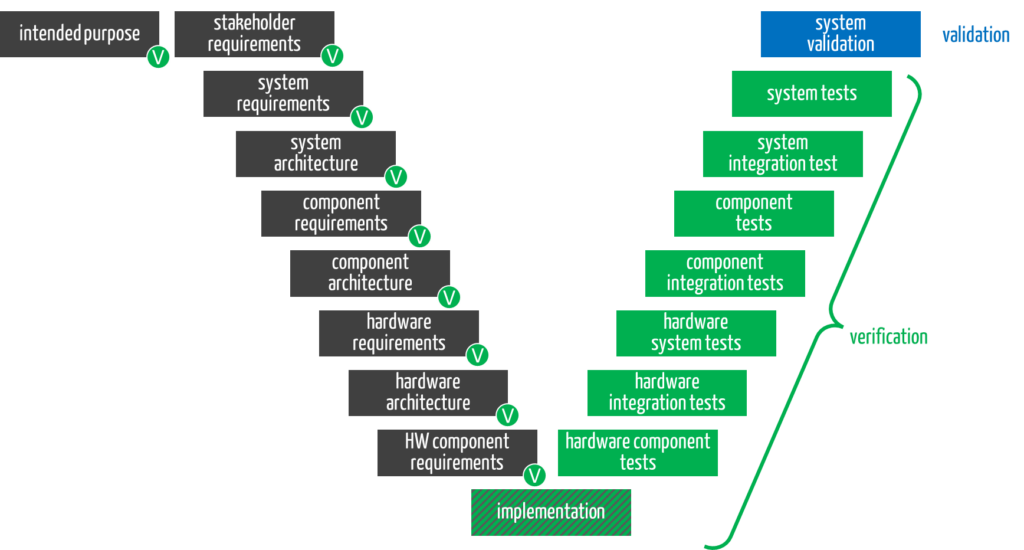
The development documents created during the design phase (gray boxes) must also be verified (green circles). The validation of the overall system completes the development (blue box).
5. Conclusion
Successful and legally compliant verification and validation are among the most important prerequisites for the approval of medical devices. Therefore, manufacturers should not make any mistakes, not even in the use of terms.
To reduce troubles, we provide various support individually tailored to your needs:
- The free starter kit provides a rapid overview of the regulatory landscape and leads to the “approval” of your medical device in six steps.
- A short article on software system requirements explains how to formulate them and thus create the prerequisites for software system tests.
- Another article describes what needs to be considered during the clinical evaluation of Medical Device Software (MDSW).
- In our usability labs, we evaluate the usability of your products (verification and validation).
Change history
- 2024-04-14: Link to pre-print removed, link to publication moved, conclusion inserted, editorial changes
- 2023-04-18: V-model added
- 2023-02-18: Publication on verification and validation linked
- 2021-09: Article completely revised, restructured, regulatory requirements updated, video replaced


